Introduction
The World Health Organisation (WHO) classification categorises soft tissue tumours (STT) based on their similarity to normal adult tissue. Namely, each STT is categorised as adipocytic, fibroblastic/myofibroblastic (F/M), so-called fibrohistiocytic, smooth-muscle, pericytic, vascular, skeletal-muscle, nerve sheath, gastrointestinal stromal tumours, and tumours of uncertain differentiation [1]. The most recent WHO classification, edited in 2013, integrates novel genetic data and provides an updated classification scheme that integrates biological behaviour as a distinguishing feature in each subcategory; STTs are further sub-grouped as benign, intermediate (locally aggressive or rarely metastasizing), and malignant [2].
The vast majority of clinically encountered STTs are benign with an excellent prognosis after curative surgical excision. Only a small proportion of STTs are malignant, although they require a multidisciplinary approach preferably at a sarcoma centre for optimal outcome. Although malignant STT or soft tissue sarcomas (STS) are infrequent in routine radiology practice, musculoskeletal radiologists must be familiar with their imaging appearance to initiate the referral to an orthopaedic oncologist or tertiary referral centre. Radiologists play an important role in the initial characterisation of STTs, with careful analysis of their size, location, tissue layer (relationship or depth relative to the fascia), and overall anatomic extent including extra-compartmental extension and neurovascular invasion. Magnetic resonance imaging (MRI) is the ideal modality for the detection, characterisation, and local staging of STT. This review will discuss the most recent updates to the WHO classification of STT that are relevant to radiologists in a clinical practice with MRI correlation. The role of advanced MRI sequences such as diffusion-weighted imaging (DWI), dynamic contrast enhanced sequences (DCE), and MR spectroscopy (MRS) are highlighted.
Adipocytic tumours
Of all the STT, adipocytic tumours are the most common mesenchymal tumours [3]. The most recent edition of the WHO Classification of tumours of soft tissues has provided several updates to the adipocytic tumour category. In the benign adipocytic tumour sub-group, the updates include removal of myxolipoma as a diagnosis because these are now considered spindle cell variants. In addition, diffuse lipoblastoma is now the preferred term for lipoblastomatosis. Of the benign adipocytic STT, spindle cell/pleomorphic lipoma can be challenging to accurately diagnose on imaging due to variable imaging appearances, lack of macroscopic fat on MRI or computed tomography (CT), and an overlapping imaging appearance with STS [4]. As such, although a simple lipoma can be diagnosed definitively on MRI, spindle cell lipoma can be a diagnostic dilemma and requires a biopsy for definitive diagnosis. As of now, there are no advanced MRI sequences addressing this challenge.
For the intermediate (locally aggressive) category of adipocytic tumours, atypical lipomatous tumour (ALT) is now divided into three main subgroups (adipocytic [lipoma- like], sclerosing, and inflammatory types) rather than four, as in the past. Although the term spindle cell liposarcoma still exists in the text, the absence of MDM2 immunopositivity or 12q15 amplification in this tumour type suggests spindle cell liposarcoma and represents a separate group of STS. MDM2 and CDK4 immunohistochemistry and/or fluorescence in situ hybridisation for MDM2 gene amplification have come into widespread use for the diagnosis of ALT/well-differentiated liposarcoma (WDLPS) and dedifferentiated liposarcoma (DDLPS) [5]. Although ALT is the preferred term, the term WDLPS is used for a histologically identical adipocytic tumour situated in anatomic locations where complete resection is not feasible, such as the mediastinum and retroperitoneum. A complete resection of a mediastinal or retroperitoneal WDLPS can be associated with significant morbidity by locally aggressive growth, recurrence, and extensive surgical resections that may require removal of multiple organs, while resection of a histologically identical tumour in the extremities (ALT) is less morbid. Using routine non-contrast and contrast-enhanced routine MRI sequences (Figure 1), lesion size, location (deep to the superficial fascia), proximal lower limb lesions, incomplete fat suppression, or increased architectural complexity (thick septations > 2 mm and nodularity > 1 cm) have been shown to be independently predictive of ALT rather than benign lipoma [6]. There is no utility of advanced MRI sequences such as DWI for the distinction of benign, intermediate, and malignant adipocytic STT, and limited added value of DCE distinguish DDLPS from ALT [7], potentially providing a means of triaging patients toward follow-up rather than biopsy in lesions with high-grade features.
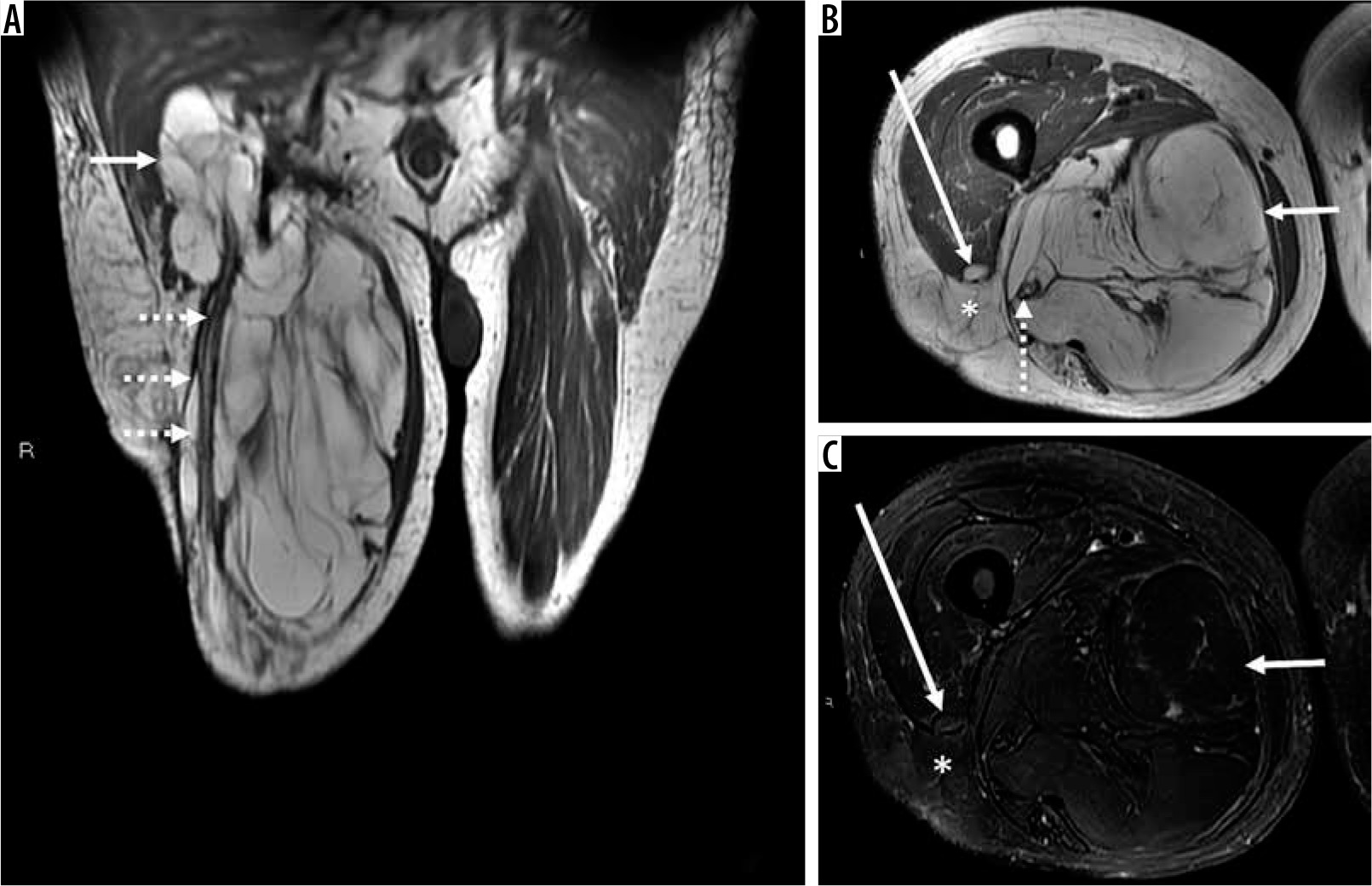
Figure 1
86-year-old man with atypical lipomatous tumour. There is a 13.2 × 12.9 × 31.7 cm recurrent macroscopic fat-containing mass occupying the medial and posterior compartments of the right thigh visible on coronal (A) and axial (B) T1-weighted and fat-suppressed T2-weighted (C) magnetic resonance (MR) images. Although there is suppression of macroscopic fat on axial T2-weighted fat-suppressed MR image through the right thigh (C), MRI features concerning for atypical lipomatous tumour (ALT) including multiple thick septations, deep location, and large size are noted. Proximally and distally, the mass surrounds the sciatic nerve (dotted arrows on images A and B). There is a small defect in the right vastus lateralis muscle with intervening fat and superficial fascia with extra-compartmental extension into the subcutaneous tissues (denoted by an * on images B and C and anterior thigh compartment (long arrows on images B and C. On routine non-contrast and contrast-enhanced routine MRI sequences, lesion size, tissue layer (deep to the superficial fascia), location (proximal lower limb), incomplete fat suppression, or increased architectural complexity (thick septations > 2 mm and nodularity > 1 cm) have been shown to be independently predictive of ALT rather than benign lipoma
Malignant adipocytic tumours (including DDLPS, myxoid liposarcoma, pleomorphic liposarcoma, and liposarcoma not otherwise specified) can be readily distinguished from both benign lipomas and ALTs due to the absence of significant intratumoural macroscopic fat. The term DDLPS is a new addition and refers to a tumour in which the high-grade or differentiated component of the tumour has adipocytic features similar to pleomorphic liposarcoma (referred to as homologous lipoblastic differentiation or pleomorphic liposarcoma-like features).
Fibroblastic/myofibroblastic tumours
The 2013 WHO classification has made several changes in the fibroblastic/myofibroblastic (F/M) tumour category. Among the benign F/M tumours, nodular fasciitis, previously considered a reactive process, is now definitively established as a self-limiting neoplasm associated with rapid growth and ultimately spontaneous regression over the course of months based on MYH9-USP6 gene fusion [8]. On histology, some cases may be worrisome for sarcoma, especially if biopsied during the active growth phase when abundant mitotic activity is present. On MRI, both proliferative myositis and nodular fasciitis appear as homogeneous masses with hypointensity or isointensity to skeletal muscle on anatomic images and as moderate or marked hyperintensity relative to skeletal muscle on T2-weighted images [9]. After administration of intravenous contrast (IV) material, nodular fasciitis tends to have homogeneous enhancement but can be variable [10]. Although the presence of a fascial tail [10] and close proximity to the superficial fascia are useful diagnostic clues, they are not determinate MRI features and histological confirmation is required [11]. Anatomic location is particularly useful in the definitive diagnosis of several benign F/M tumours including elastofibroma (Figure 2), fibromatosis colli, and fibroma of tendon sheath.
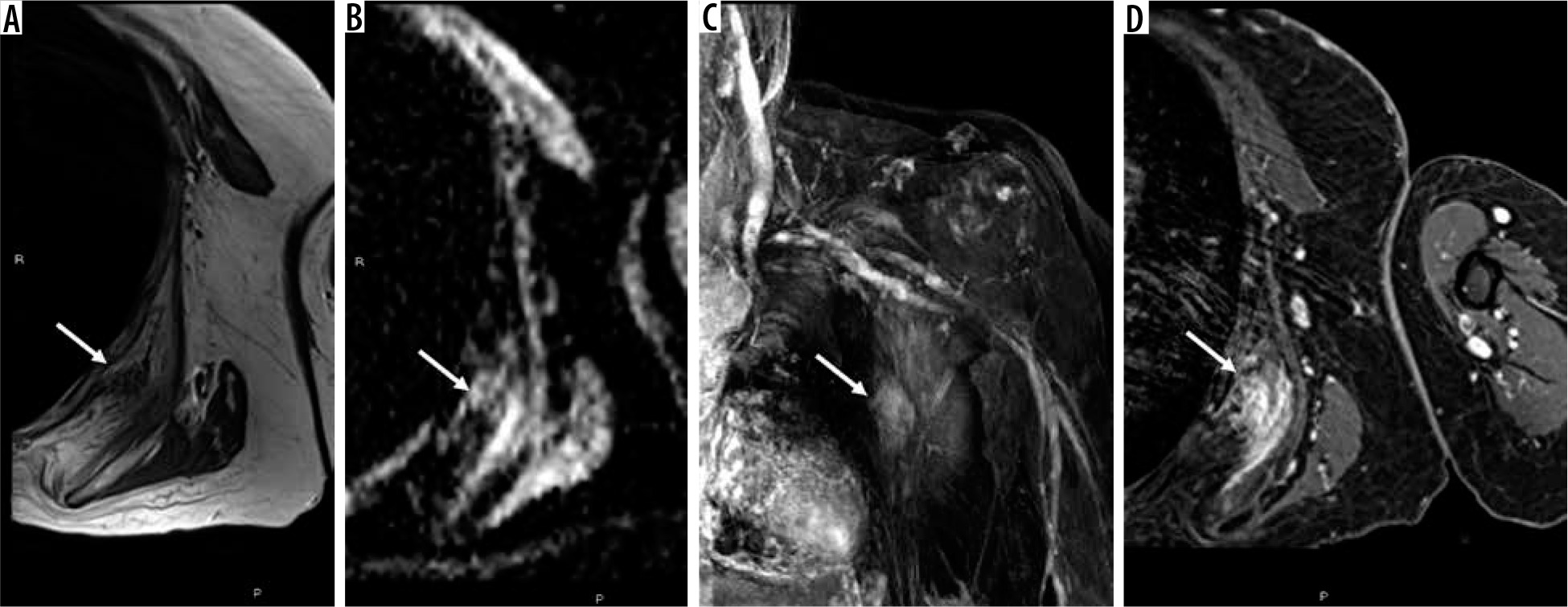
Figure 2
67-year-old woman with elastofibroma dorsi. Axial T1-weighted magnetic resonance (MR) image through the left chest wall (A) shows an unencapsulated mass-like abnormality deep to the scapula (arrow) with corresponding indistinct elevated signal on ADC map (B), late arterial enhancement on coronal DCE MIP (C), and confluent enhancement on static T1-weighted fat-suppressed post-contrast axial MR image (D). The mass was biopsied and confirmed to be elastofibroma
Of the benign STTs in this category, myositis ossificans (MO) remains a diagnostic dilemma because it is frequently mistaken for a sarcoma and biopsied (Figure 3). Although MO is typically considered a non-neoplastic, post-traumatic entity, it is included as in the F/M tumour category based on the frequent presence of USP6 rearrangement, similar to nodular fasciitis [12]. On MRI, MO has a non-specific appearance in its early (< 4 weeks) and intermediate (4-8 weeks) imaging as a heterogeneous, intramuscular mass that is isointense or slightly hyperintense on T1-weighted imaging and hyperintense on T2-weighted imaging, with diffuse and avid enhancement after the administration of IV contrast material, and extensive soft tissue oedema [13]. The presence of extensive perilesional muscle oedema as a useful imaging feature for the distinction of early/intermediate stage MO from STS offers moderate sensitivity of 60-70% and excellent specificity of 97-99% [13]. Faint peripheral calcification is visible in the early stage, which progresses to variable thickness calcification in the intermediate stage. The combination of typical MRI features in STT measuring less than 5 cm, extensive perilesional oedema, history of trauma, and faint peripheral zonal calcification can be diagnostic of MO and followed closely on imaging [13].
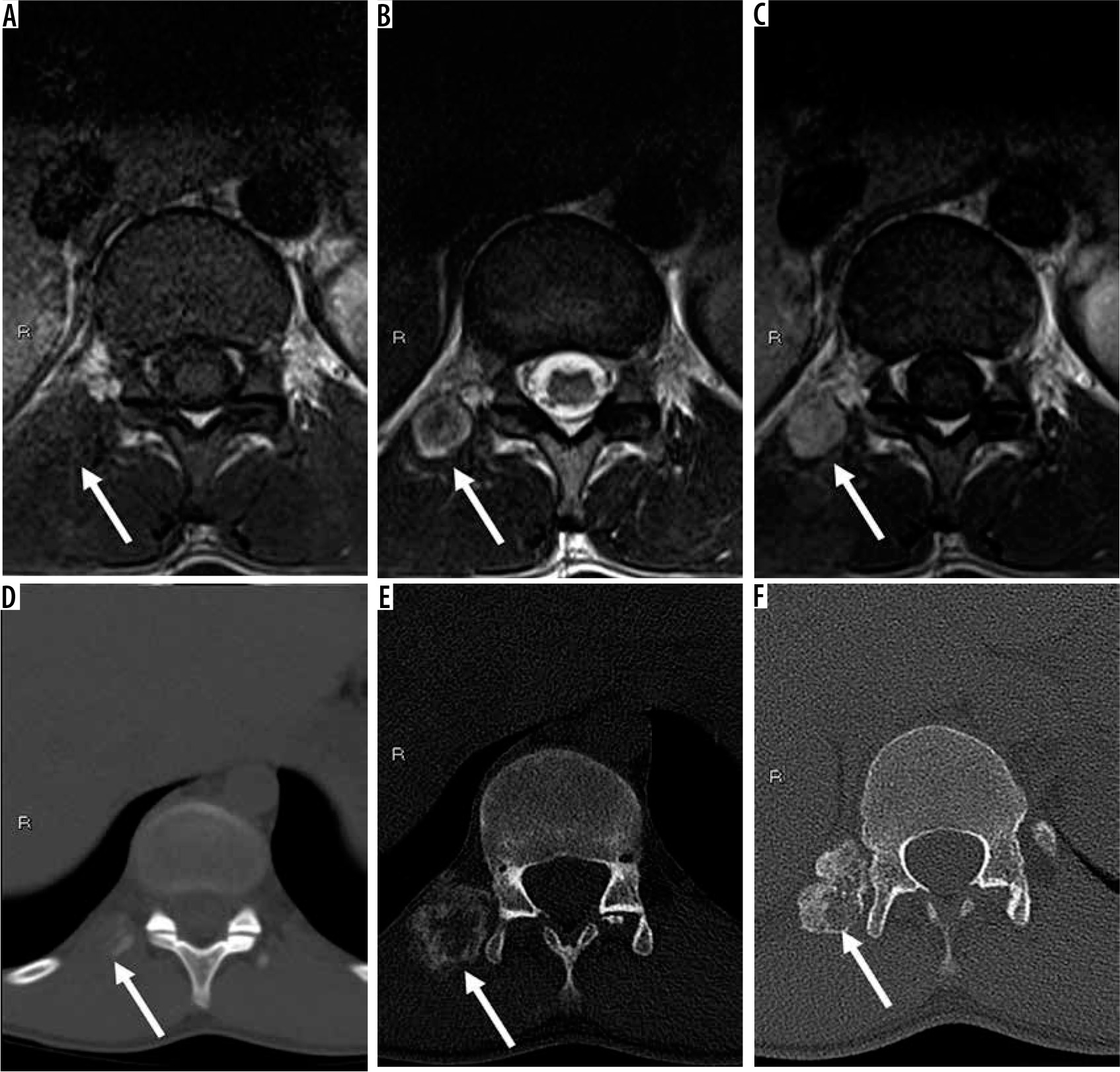
Figure 3
20-year-old man with right paraspinal mass. Axial T1-weighted (A), T2-weighted (B), and T1-weighted post-contrast (C) magnetic resonance (MR) images through the thoracolumbar junction show a right paraspinal soft tissue mass (arrows) that was initially characterised as an indeterminate soft tissue mass suspicious for peripheral nerve sheath tumours (PNSTs) based on target sign visible on T2-weighted image (B) and paraspinal location. The patient underwent a biopsy, which was interpreted as a malignant PNST at an outside facility. The histological slides were re-interpreted as myositis ossificans (MO), cellular phase. Sequential axial computed tomography images through the lower thoracic spine at the time of initial presentation (D), in the intermediate stage (E), and late stage (F) reveal progressive zonal ossification peripherally compatible with MO (arrows). Although the presence of extensive perilesional muscle oedema is a useful imaging feature for the distinction of early/intermediate stage MO from sarcoma, it is not always visible, as in this case
In the intermediate (locally aggressive) sub-group, palmar/plantar fibromatosis can also be diagnosed definitively based on its anatomic location. Palmar/plantar fibromatosis (Figure 4) demonstrates multiple nodular or band-like soft tissue masses arising from the proximal palmar or plantar aponeurosis extending along the subcutaneous tissues with variable T1 and T2 signal intensities with associated heterogeneous enhancement depending on the content of collagen and cellularity, similar to the other fibromatoses [14]. Plantar fibromatosis manifests as a small cluster of multiple tumours along the deep plantar aponeurosis, which typically blend with the adjacent plantar musculature. In the setting of a solitary tumour, the term plantar fibroma rather than fibromatosis may be used. Fascial tails along the aponeurosis are frequent.
Figure 4
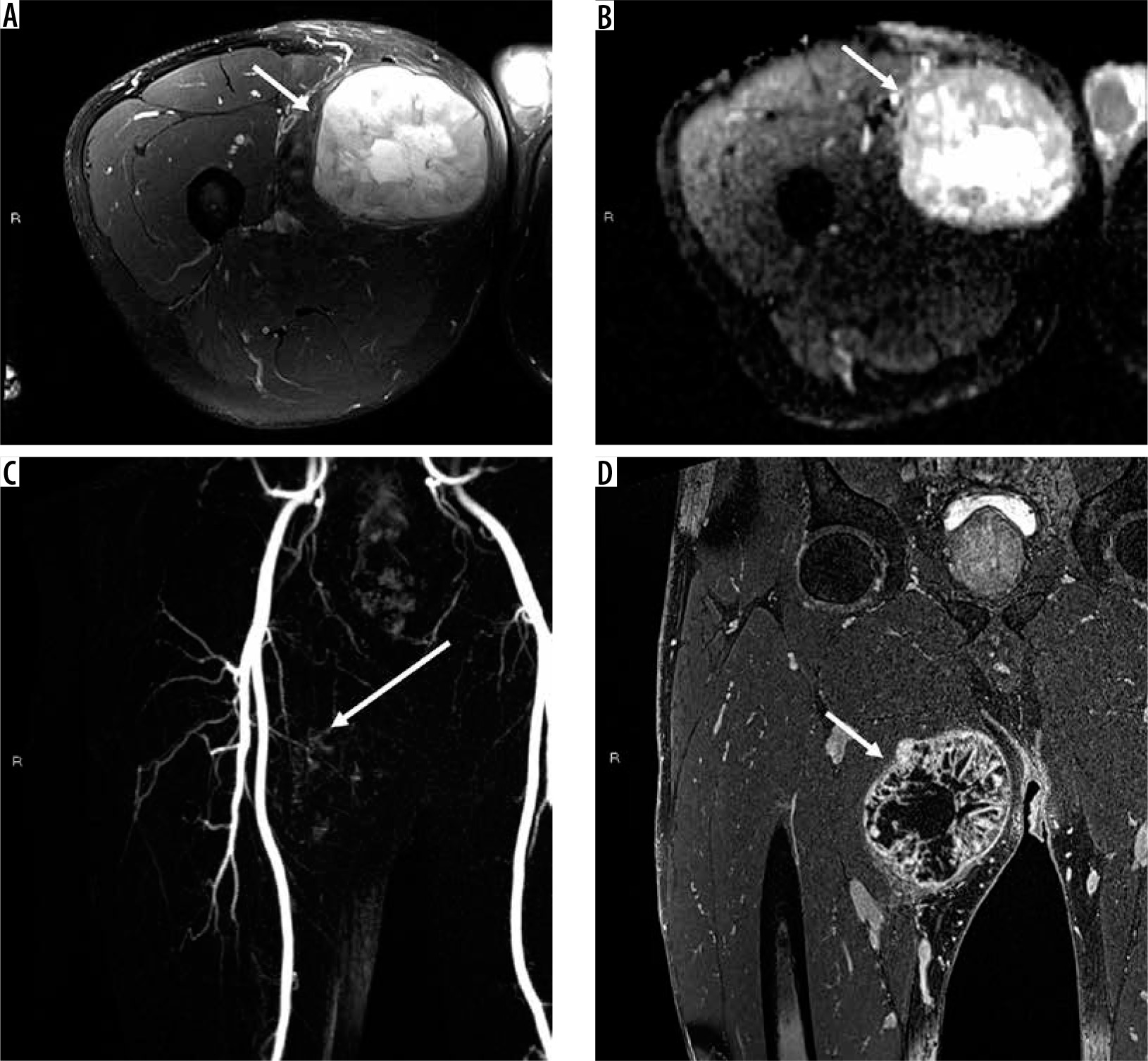
32-year-old man with palmar fibromatosis. There is an unencapsulated, nodular soft tissue mass arising from the proximal palmar aponeurosis extending along the subcutaneous tissues (arrows) that is isointense to skeletal muscle on axial T1-weighted (A), hyperintense to skeletal muscle on axial T2-weighted fat-suppressed (B) with confluent internal enhancement on axial (C) and sagittal (D) T1-weighted fat-suppressed subtraction magnetic resonance (MR) images through the hand. Fascial tails along the aponeurosis are visible (dotted arrow on image D). Coronal dynamic MR angiogram image (E) shows late arterial enhancement, a feature of benign disease
Desmoids-type fibromatosis (Figure 5) also exhibits similar signal characteristics with fascial tails but occurs in the trunk. Desmoids often have band-like low signal internally, which can suggest the diagnosis, although this entity can be diagnostically challenging and require a biopsy prior to definitive treatment [14]. Two new entities, giant cell fibroblastoma and dermatofibrosarcoma protuberans (DFSP), which were previously classified as skin tumour neoplasms, are new additions in the intermediate (rarely metastasising) sub-group. Giant cell fibroblastoma shows frequent local recurrence but has no metastatic potential. DFSP, on the other hand, shows not only locally aggressive behaviour but can also rarely transform into a fibrosarcoma and metastasise. DFSP tends to be subcutaneous in location with nonspecific T1 and T2 signal characteristics (Figure 6). Differential diagnosis of an aggressive subcutaneous STT includes DFSP, undifferentiated pleomorphic sarcoma (UPS)/fibrosarcoma, cutaneous/subcutaneous neoplasms (melanoma) and metastatic disease [14].
Figure 5
40-year-old woman with desmoid type fibromatosis. Desmoids-type fibromatosis exhibits similar signal characteristics to palmar/plantar fibromatosis with fascial tails but tends to occur in the trunk. Axial T1-weighted magnetic resonance image through the right anterior chest wall (A) shows an unencapsulated mass at the costochondral junction (arrow) with a corresponding indistinct low signal on T2-weighted image (B), and confluent enhancement on static T1-weighted fat-suppressed post-contrast axial image (C). Fascial tails (dotted arrows) along the chest wall are visible
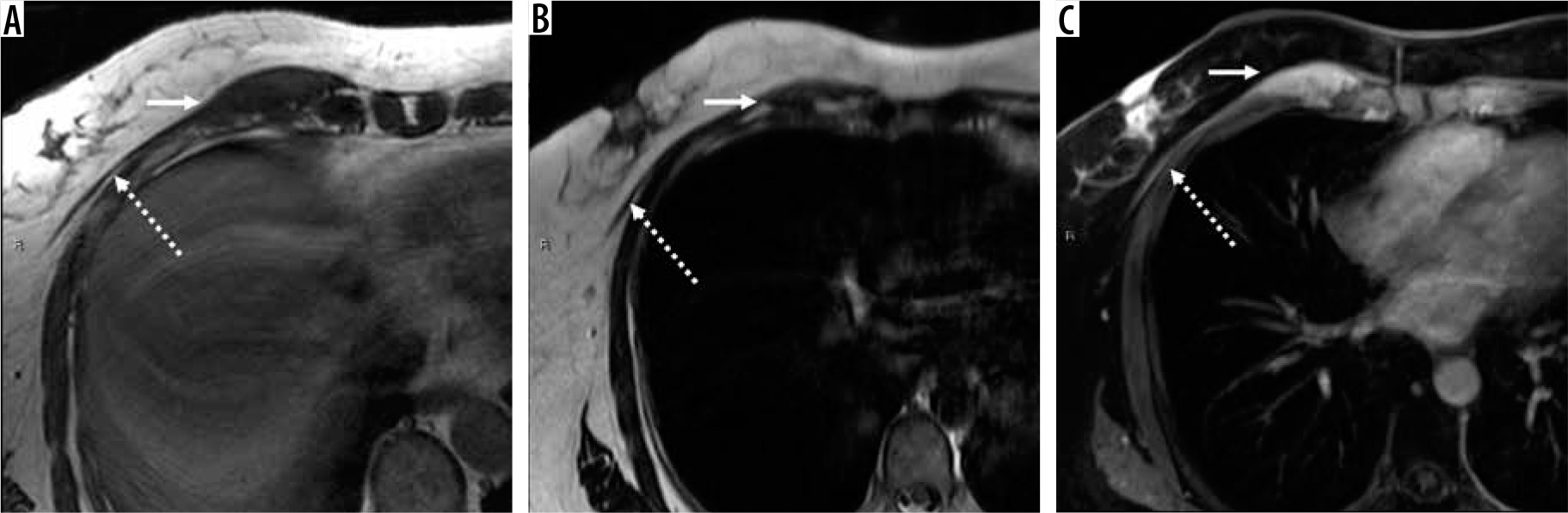
Figure 6
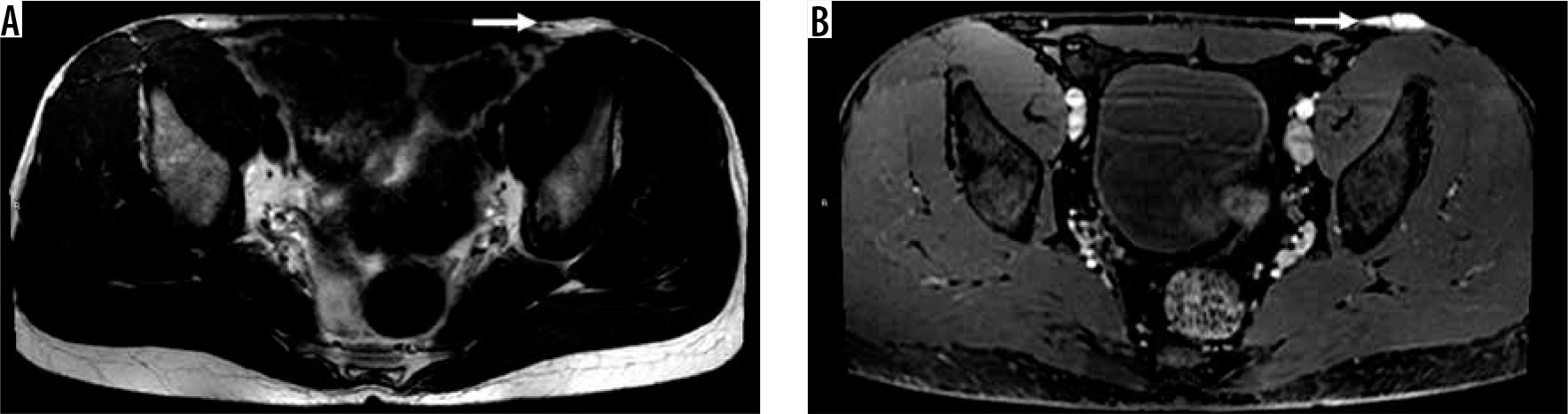
35-year-old male with dermatofibrosarcoma protuberans (DFSP). There is a 4.8 × 2.0 × 0.9 cm multilobulated T2 hyperintense (A), homogeneously enhancing dermal-based lesion (arrow) on axial T1-weighted post-contrast fat-suppressed image (B) through the pelvis located in the region of the left inguinal crease, with plaque-like thickening of the skin. DFSP, previously classified as a skin tumour, is a new addition in the intermediate (rarely metastasising) sub-group of fibroblastic/myofibroblastic soft tissue tumours
Lastly, myofibroma/myofibromatosis are now described under the categories of pericytic tumours and solitary fibrous tumour, haemangiopericytoma, lipomatous haemangiopericytoma, and giant cell angiofibroma and are all re-categorised as solitary fibrous tumours (SFT). On MRI, SFTs are nonspecific, well-defined, ovoid, and heterogeneously enhancing with regions of intra-lesional low signal intensity on fluid-sensitive sequences that correspond to dense collagen and a vascular pedicle [15,16]. On DCE-MRI, the fibrous variety of SFTs tends to differ from its cellular counterparts, in that fibrous SFTs show progressive enhancement following contrast administration while cellular SFTs exhibit a rapid initial enhancement pattern. On DWI, however, there is an overlap in the mean ADC values (× 10-3 mm2/s) of cellular and fibrous SFTs (1.4 ± 0.4 vs. 1.4 ± 0.5, respectively) [17]. There are no data regarding the utility of minimum ADC values in distinguishing these two kinds of SFT. There are no determinate features of malignant F/M STT; however, the presence of restricted diffusion on DWI/ADC mapping and early arterial enhancement on DCE are indicators of malignancy (Figure 7).
Figure 7
66-year-old man with malignant fibroblastic/myofibroblastic tumour. Axial T2-weighted fat-suppressed magnetic resonance image through the right proximal thigh (A) shows an encapsulated 12 cm heterogeneous centrally higher signal intensity soft tissue mass located deep to the superficial fascia (arrow) with corresponding low-signal/restricted diffusion on ADC map (B), early arterial enhancement on coronal DCE MIP (C), and heterogeneous enhancement with intralesional necrosis on static coronal T1-weighted fat-suppressed post-contrast coronal image (D). The mass was biopsied and con- firmed to be grade 2 myxofibrosarcoma
So-called fibrohistiocytic tumours
Most commonly encountered so-called fibrohistiocytic tumours are benign and include tenosynovial giant cell tumour (TSGCT) (localised type, diffuse type, and malignant) as well as deep benign fibrous histiocytoma. Fibroma of tendon sheath, a benign F/M tumour, can mimic a TSGCT. Both neoplasms can present clinically as indolent, slow-growing, painless masses located in close proximity to tendon sheaths, typically of the extremities. On MRI, both neoplasms tend to be low or variable signal intensity on non-contrast anatomic and fluid sensitive sequences, with little to no enhancement following contrast administration. Gradient echo (GRE) images can be useful to diagnose TSGCT based on the presence of susceptibility artifact (hemosiderin deposition). By other sequences, fibroma of tendon sheath appears as a nodular and mass-like lesion with a strip-like or irregular signal characteristics on fluid-sensitive sequence compared with the casting, mould morphology, and flaky/granular or separated pattern of TSGCT [18]. The latter distinguishing MRI features offer high sensitivities (83-100%) and diagnostic accuracies (60-89%) but slightly lower specificities (29-79%) [18].
Intermediate (rarely metastasising) so-called fibrohistiocytic tumours include plexiform fibrohistiocytic tumour and giant cell tumour of soft tissue [1]. Malignant so-called fibrohistiocytic tumours, formerly known as malignant fibrous histiocytoma (MFH), and its subtypes have been renamed as undifferentiated sarcoma and are now classified under the undifferentiated/unclassified sarcomas section described below [2].
Smooth muscle tumours
The only change in this group has been the removal of angioleiomyoma, which is now included in the category of pericytic tumours [2].
Pericytic (perivascular) tumours
Pericytic (perivascular) tumours include glomus tumour and its variants as well as glomangiomatosis and, rarely, malignant glomus tumour. The 2013 WHO classification revised recommendations for the designation of glomus tumours as malignant and of uncertain malignant potential. The term malignant glomus tumours is used in the setting of marked nuclear atypia and any mitotic activity [2]. Tumour size greater than 2.0 cm and deep location are no longer criteria for malignancy, and tumours with these features but no nuclear atypia are now considered to be glomus tumours of uncertain malignant potential [2,19]. Glomus tumours can also be associated with Neurofibromatosis type 1 (NF1) and Horner syndrome. Although the majority of glomus tumours (65-75%) occur in the hand, with the most common location being the tip of the finger or toe, extra-digital glomus tumours also occur and present clinically as painful masses [19]. On MRI, the appearance of glomus tumours overlaps with other vascular neoplasms [20]. An additional major update in this category includes reclassification of myofibroma and myofibromatosis under myopericytoma rather than F/M tumours [2].
Skeletal muscle tumours
Although there have been modifications to the skeletal muscle tumour category based on advances in genetic mutations associated different types of rhabdomyosarcoma, advanced MRI sequences have not provided similar insight into tumour category, and there are no diagnostic imaging features that enable diagnostic certainty without a biopsy.
Gastrointestinal stromal tumours
Gastrointestinal stromal tumours (GISTs) were previously categorised under tumours of the digestive system but are now under the soft tissue tumour classification. GISTs can be benign, of uncertain malignant potential, and malignant. The widely accepted National Comprehensive Cancer Network guidelines for risk stratification of GISTs are based on anatomic site, tumour size, and mitotic count, features determined to be highly predictive of malignant behaviour in several large retrospective studies [21,22]. Several qualitative and quantitative MRI and 18F-fluorodeoxyglucose positron emission tomography – computed tomography (18F-FDG PET/CT) features including ADC and maximum standard uptakes values (SUVmax) values can be useful in distinguishing non-GISTs, low-risk GISTs, and high-risk GISTs [23]. On multivariate analysis, signal characteristics on T2-weighted imaging was the only independent variable for differentiating GISTs from non-GISTs (such as leiomyoma, schwannoma, and glomus tumour) [23]. Enhancement characteristics, mean ADC values, and SUVmax have been found to be useful features for differentiating between high-risk and low-risk GISTs [23]; high-risk GISTs tend to have lower mean ADC values and higher SUVmax when compared with low-risk GISTs and non-GISTs [23]. The utility of minimum ADC values, as opposed to mean ADC values, has not been investigated.
Vascular tumours
Vascular STT are classified as benign (haemangioma and epithelioid haemangioma), intermediate/locally aggressive (kaposiform haemangioendothelioma), intermediate/rarely metastasising (haemangioendothelioma and Kaposi sarcoma), and malignant (epithelioid haemangioendothelioma and angiosarcoma of soft tissue). Two new entities, pseudomyogenic haemangioendothelioma (and also known as epithelioid sarcoma-like haemangioendothelioma), have been added to the intermediate (rarely metastasising) vascular tumour category. Pseudomyogenic haemangioendothelioma tends to be very avid on 18F-FDG PET/CT, which can be useful for the detection of deep lesions [24]. The clinical course of patients with pseudomyogenic haemangioendothelioma is characterised by repeated local recurrence or the development of new tumour nodules within the same anatomical region, but metastasis is very rare [2]. The relationship between margin status and risk of disease recurrence has not been established, and at this point conservative management is the treatment of choice [25]. Although there have been new insights into the molecular genetics of malignant vascular tumours, the classification of de novo as well post-radiation angiosarcoma and epithelioid haemangioendothelioma and its implications for radiologists has remained the same [2].
Nerve sheath tumours
Peripheral nerve sheath tumours (PNSTs) are included in the WHO soft tissue tumour classification rather than tumours of the central nervous system. Although several newly described histological variants of benign PNSTs have been included, there have been no changes to the classification of malignant peripheral nerve sheath tumours. Benign PNSTs include schwannoma (and its variants), melanotic schwannoma, neurofibroma (and its variants), plexiform neurofibroma, perineurioma, granular cell tumours, dermal nerve sheath myxoma, ectopic meningioma, nasal glial heterotopia, benign triton tumour, and lastly hybrid nerve sheath tumours (which have features of more than 1 type of conventional PNSTs). The most common hybrid lesions are a hybrid schwannoma and perineurioma, which is typically sporadic, and a hybrid neurofibroma/schwannoma, which is associated with type 1 neurofibromatosis [26].
Of the various STTs, advanced MRI sequences such as DWI and ADC mapping have been particularly useful for the distinction of benign PNSTs from malignant PNSTs. First, a morphological target sign visible on fluid sensitive and contrast-enhanced MRI sequences is indicative of a benign PNST (both schwannoma and neurofibroma) [27] and rarely presents in a malignant peripheral nerve tumour. The qualitative presence of target sign (Figure 8) is more often seen on DWI using high b-values and ADC mapping based on the intrinsic T2 properties of a PNST compared with routine MRI sequence, and suggests the preservation of the PNSTs internal histological architecture [27]. In addition, quantitative ADC values have also been used as a biomarker for malignancy in people with PNSTs [28,29] including nerve tumour syndromes such as NF1 [30]. Mean PNST diameter (> 4.2 cm) and minimum ADC value (≤ 1.0 × 10-3 mm2/s) are potentially important parameters that may be used to distinguish malignant PNSTs from benign PNSTs with high diagnostic accuracy [28]. In people with NF1, minimum ADC value ≤ 1.0 × 10-3 mm2/s offers a high specificity of 94% while maintaining a perfect sensitivity of 100% for malignancy [30]. As such, advanced MRI sequences such as quantitative DWI are very useful in this particular subpopulation, who can have numerous PNSTs but also an inherently higher incidence of malignancy.
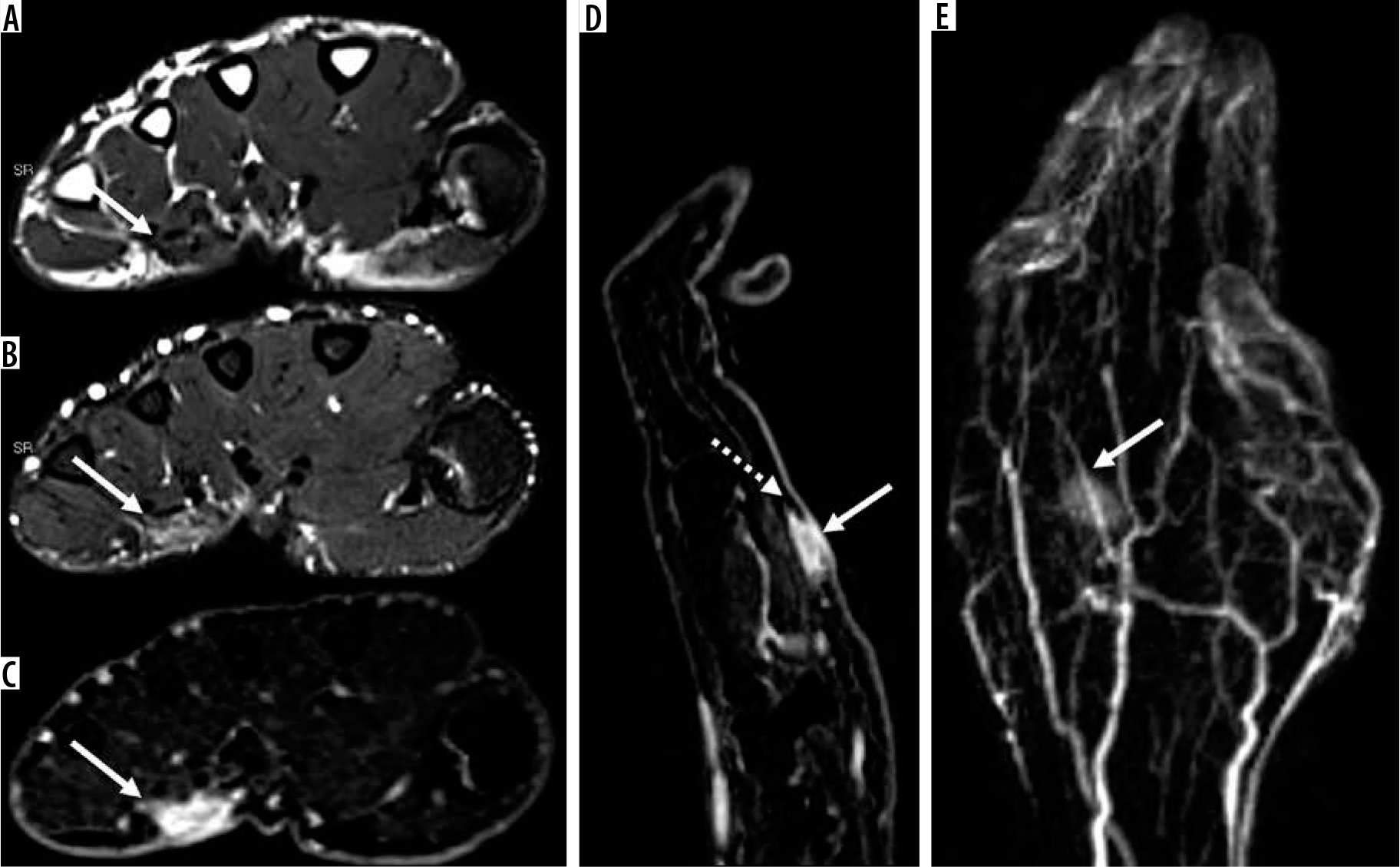
Figure 8
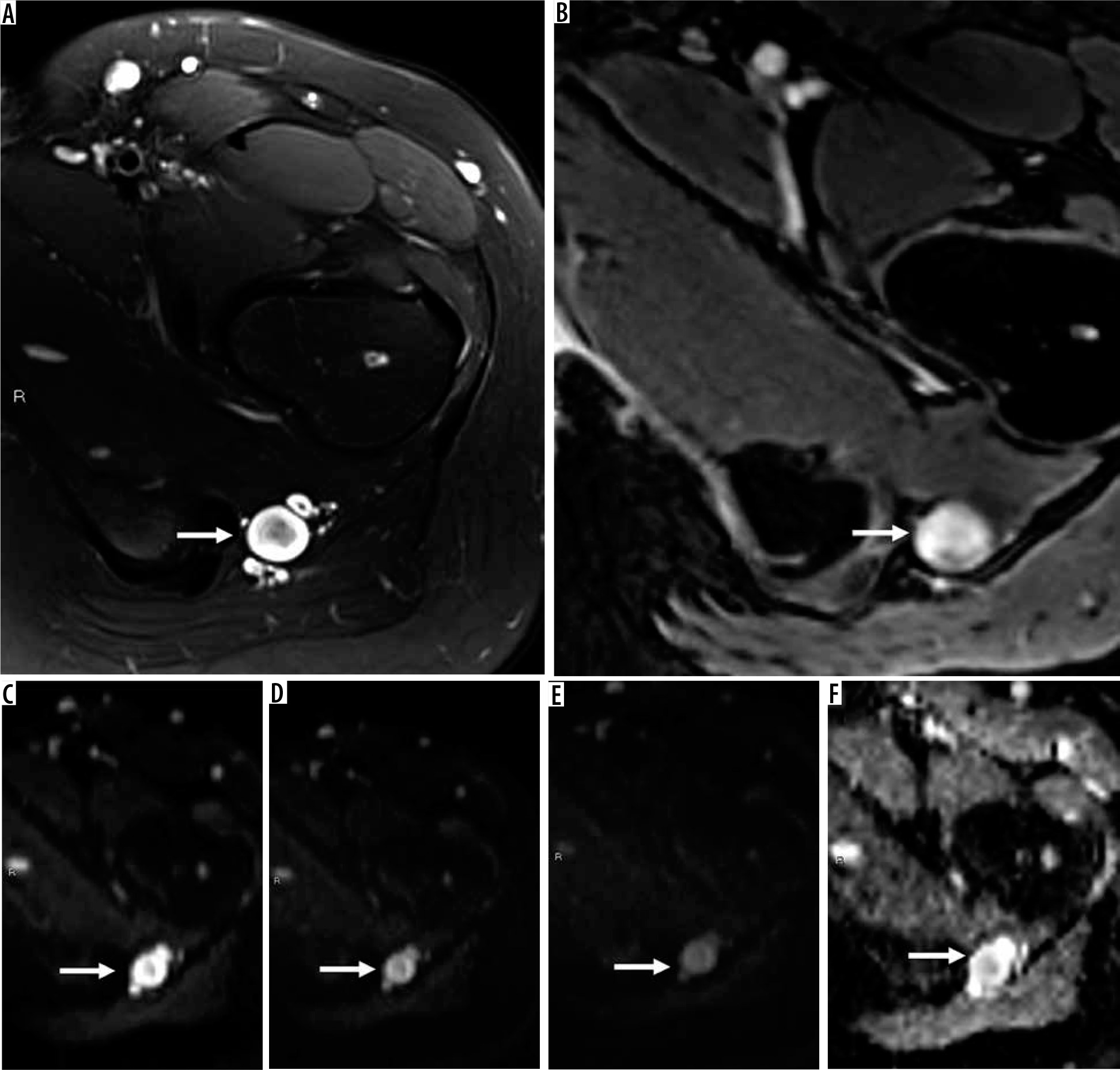
16-year-old girl with neurofibroma. Axial T2-weighted fat-suppressed (A) and T1-weighted fat-suppressed post-contrast (B) magnetic resonance( MR) images through the left proximal thigh show a small enhancing soft tissue mass along the sciatic nerve (arrows) with a target sign seen in (A). DWI using low b-value of 50 s/mm2 (C), intermediate b-value of 400 s/mm2 (D), high b-value of 800 s/mm2 (E), and lastly the ADC map (F) (arrows) show a soft tissue mass with a target sign: suggesting preserved zonal histological architecture of a peripheral nerve sheath tumours (PNST). A morphological target sign can be visible on fluid-sensitive and contrast-enhanced MRI sequences and indicates a benign PNST. The qualitative presence of target sign is more often seen on DWI using high b-values and ADC mapping based on the intrinsic T2 properties of a PNST when compared with routine MRI sequences, and suggests the preservation of the PNST’s internal histological architecture. Mean PNST diameter (> 4.2 cm) and minimum ADC values (≤ 1.0 × 10-3 mm2/s) have been found to accurate quantitative metrics that can be used to distinguish malignant PNSTs from benign PNSTs
Limited data on patients with PNSTs suggests that MRS offers a highly sensitive (100%) but non-specific (50%) metabolic method of distinguishing malignant from benign PNSTs. The added imaging and post-processing time associated with MRS in addition to slightly lower diagnostic accuracy has limited its utility in routine practice.
Tumours of uncertain differentiation
Tumours of uncertain differentiation can be benign (such as intra-muscular myxoma and pleomorphic hyalinising angiectatic tumour [PHAT]), intermediate/locally aggressive such as haemosiderotic fibrolipomatous tumour (HFLT), intermediate/rarely metastasising, which include benign and malignant varieties of phosphaturic mesenchymal tumour, and frankly malignant STT including synovial sarcoma (Figure 9), epithelioid sarcoma, alveolar soft-part sarcoma, clear cell sarcoma (Figure 10), and soft tissue extra-skeletal Ewing sarcoma as well as neoplasms with perivascular epithelioid cell differentiation.
Figure 9
17-year-old male with reported right-sided neck swelling found to be synovial sarcoma. On coronal (A) and axial (B) fluid-sensitive fat-suppressed magnetic resonance (MR) images through the neck, there is a circumscribed somewhat heterogeneous high signal intensity mass centred within the right parapharyngeal and submandibular spaces (arrow), measuring approximately 3.6 × 3.8 × 8.1 cm (AP by TR by CC). C) The mass demonstrates avid slightly heterogeneous enhancement on the T1-weighted fat-suppressed axial MR image (arrow). By routine MRI sequences, the lesion is characterised as an indeterminate solid soft tissue mass that requires biopsy
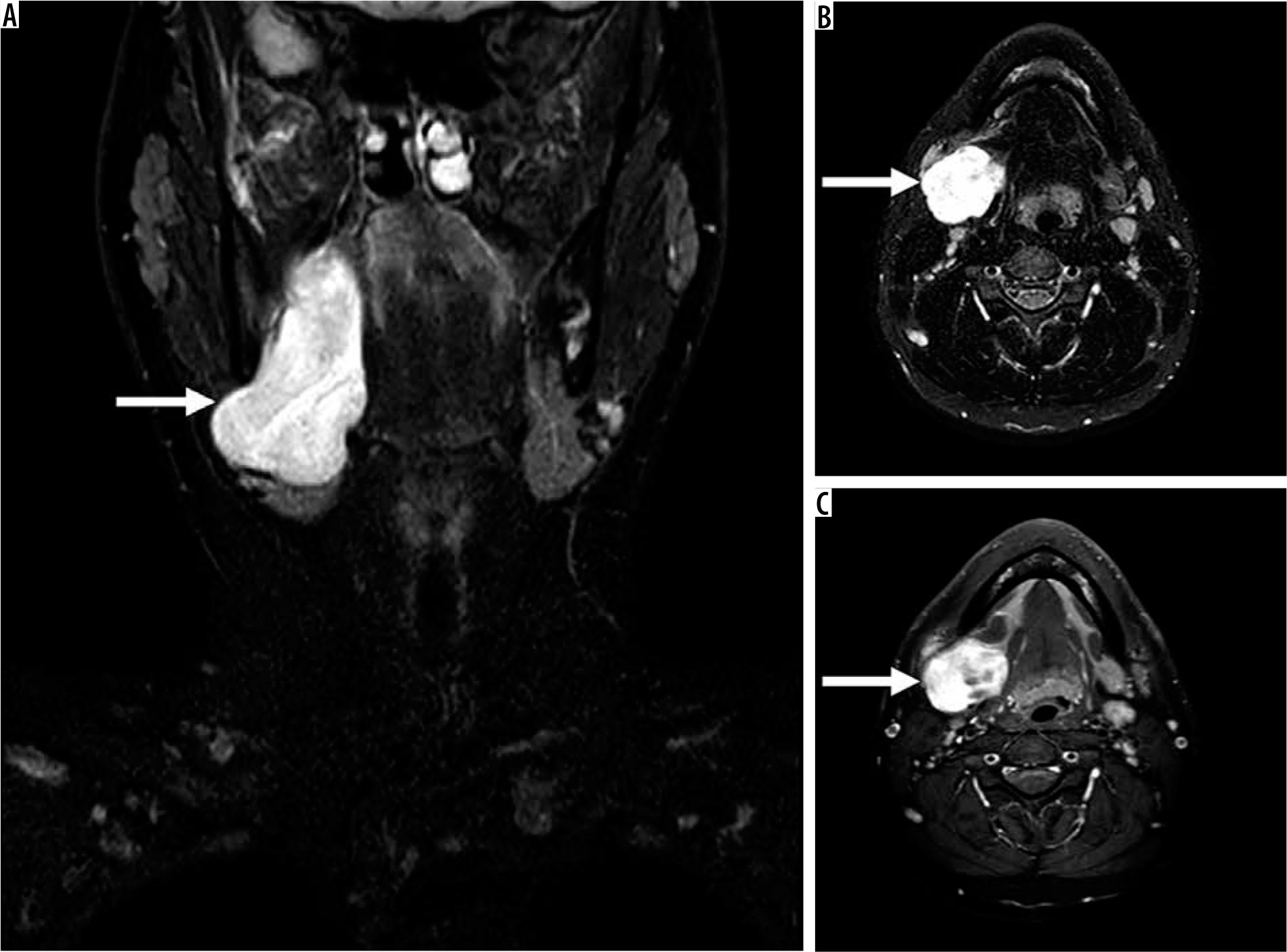
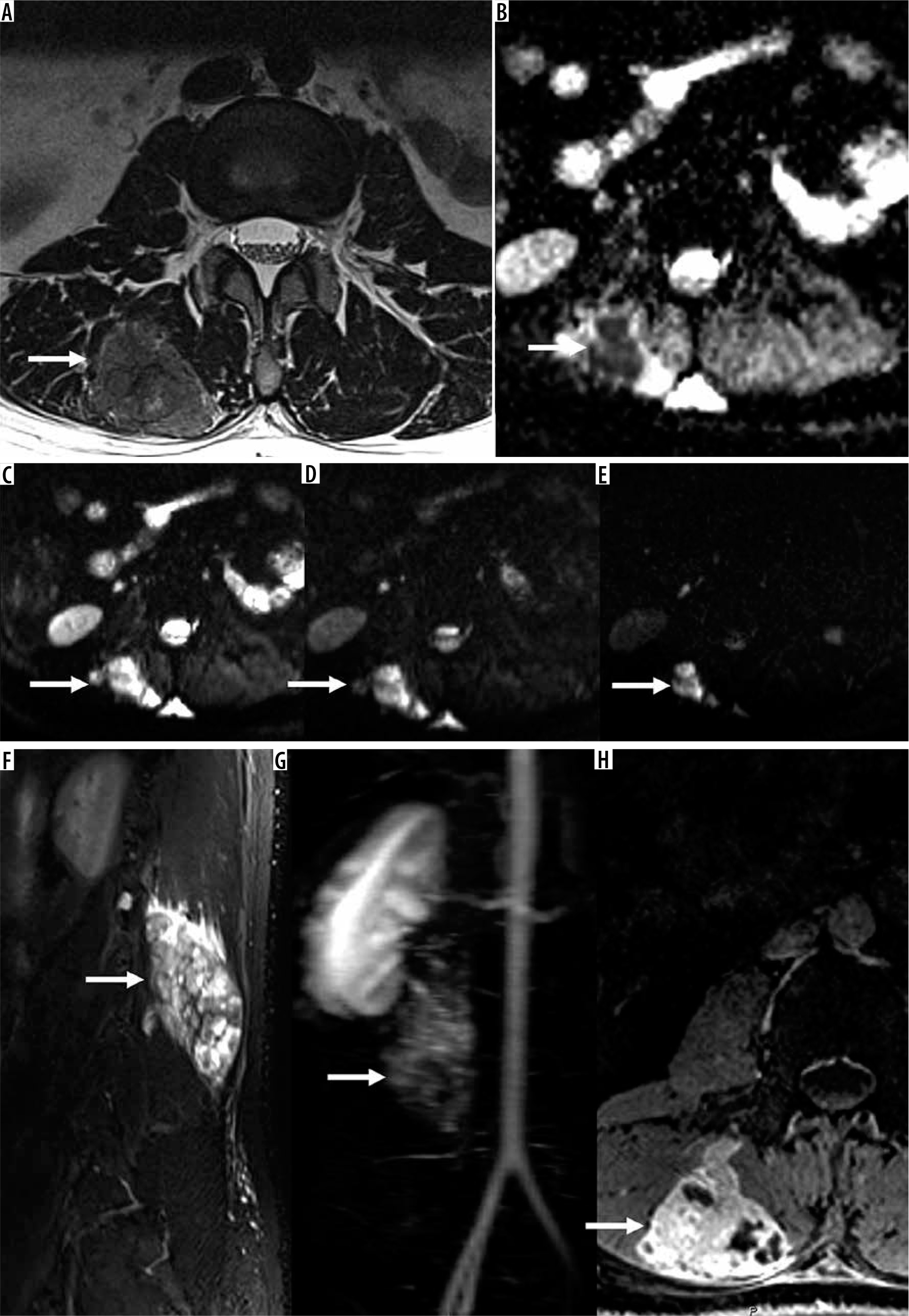
Figure 10
30-year-old woman with recurrent clear cell sarcoma. Axial T2-weighted magnetic resonance (MR) image (A) and ADC map (B) through the upper lumbar spine show a right paraspinal unencapsulated soft tissue mass (arrow) of heterogeneously low signal intensity. There is qualitative restricted diffusion on DWI using low b-value of 50 s/mm2 (C), intermediate b-value of 400 s/mm2 (D), and high b-value of 800 s/mm2 (E) with lack of signal loss on DWI with increasing b-values. On the axial ADC map (B), there is qualitative restricted diffusion or low signal and quantitatively low ADC values, compatible with malignancy. Sagittal STIR MR image (F) shows perilesional oedema and internal septations within the mass (arrow). There is early arterial enhancement on DCE MIP (G) and heterogeneous enhancement on axial T1-wighted fat-suppressed post-contrast axial MR image (H) (arrows). Features of malignancy include restrictive diffusion on DWI/ADC mapping and early arterial enhancement
Although PHATs tend to exhibit variable imaging features, a solid subcutaneous mass with ill-defined margins in the extremity should raise the possibility of PHAT [31]. Of note, these features are rather similar to other benign and malignant STTs such as nodular fasciitis. HFLT, a locally aggressive STT of uncertain differentiation typically occurring in middle-aged women, most commonly around the ankle or wrist, is an un-encapsulated, large tumour with high rate of local recurrence estimated at 50% if incompletely excised [2]. On MRI, HFLT presents as a soft tissue mass with indistinct margins, intralesional fat, and septations with local recurrence and occasionally blood products on GRE [32]. PMT, which is classified as a rarely metastasising lesion, is an extremely rare but clinically and histologically distinctive tumour that produces fibroblast growth factor 23, a hormone that blocks the reuptake of phosphate by proximal renal tubule, resulting in phosphaturia [33]. As such, PMTs produce phosphaturia and result in tumour-induced osteomalacia, which resolves with complete PMT excision [34]. Clinically, the majority of patients with PMT exhibit elevated serum levels of fibroblast growth factor 23 [33]. PMTs tend to have a non-specific imaging appearance and can mimic TSGCT, and even ALT [35]. Of note, the majority of PMTs tend to exhibit variable hypointensity on T2-weighted imaging, radiotracer uptake on 99mTc-sestamibi scintigraphy, and 111In-pentetreotide scintigraphy, as well as 68Ga-Dotate PET/CT and 18F-FDG PET/CT (mean SUVmax was 4.1 ± 2.5 with a range of 1.5-10.8) [35].
Malignant tumours of uncertain differentiation tend to have aggressive MRI features. Of these, alveolar soft-part sarcomas (ASPS) tend to exhibit eight MRI features more often: deep location relative to the superficial fascia, elevated signal on T1-weighted imaging, intralesional necrosis, lack of fibrotic component, infiltrative growth pattern, absence of tail sign, presence of intra- and perilesional flow-voids, and more than five flow voids suggesting neovascularity [36]. Although these features are not diagnostic of ASPS, their presence is compatible with the diagnosis and suggests an aggressive histology that should be referred to a sarcoma centre and worked up appropriately.
Lastly, similar to bone tumours, the term primitive neuroectodermal tumour has been removed as a synonym for Ewing sarcoma.
Undifferentiated/unclassified sarcoma
This new category recognises sarcomas that cannot be classified into any of the other categories and includes undifferentiated varieties of spindle cell, pleomorphic (Figure 11), round cell, epithelioid, and not otherwise specified STS. Undifferentiated sarcoma is distinct from de-differentiated sarcoma because sarcomas in the undifferentiated category lack clinical or morphological features that would otherwise place them under specific types of sarcomas, while dedifferentiated sarcomas tend to have some similarity to an adult tissue type. UPS was previously likely to be diagnosed as MFH. Undifferentiated/unclassified sarcomas account for up to 20% of all sarcomas, and approximately 25% of these are radiation-associated STS [2]. Similar to malignant fibroblastic myofibroblastic tumour (myxofibrosarcoma), a tail sign, defined as thick fascial enhancement extending from the sarcoma margin, is also a feature of undifferentiated sarcomas and is associated with locoregional recurrence [37]. Although there are no discrete regions of nodular enhancement along the tail sign or enhancing fascia, tumour infiltration along the fascia and subcutaneous fibrous septa is detected on histology [37]. As such, detection of the tail sign associated with a soft tissue mass is particularly important for pre-operative planning and resection of tumour margins [37]. There are no large series investigating the advanced MRI features of undifferentiated varieties of sarcoma.
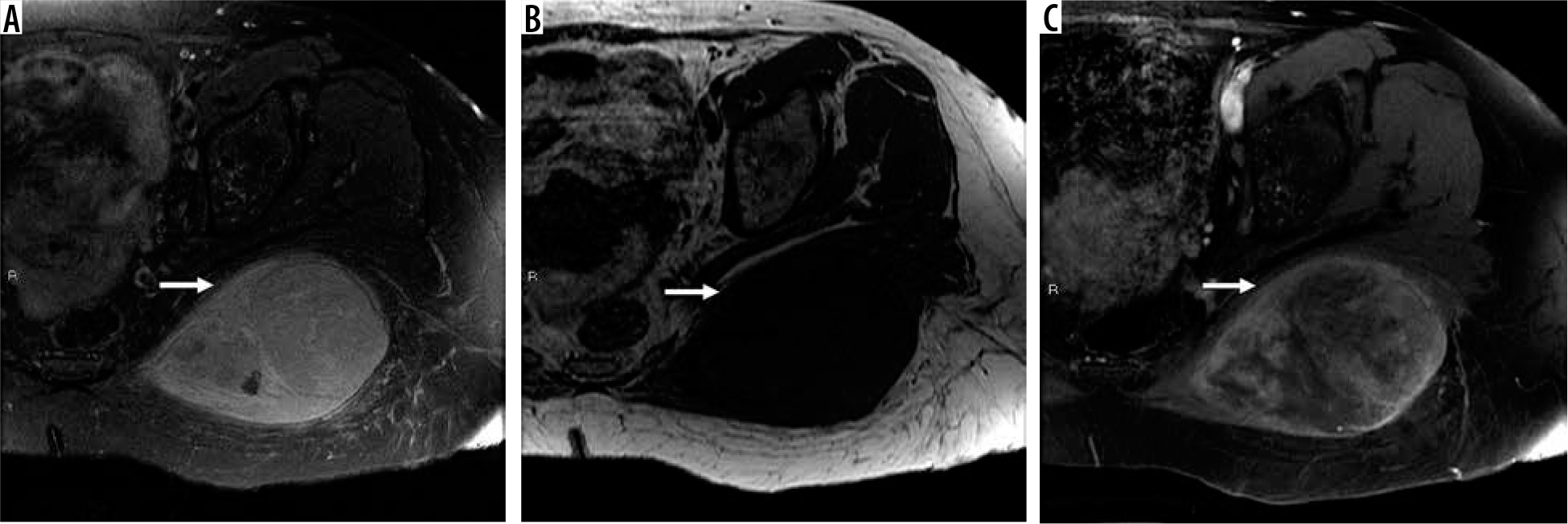
Figure 11
61-year-old woman with grade 3 undifferentiated pleomorphic sarcoma. There is a large 9.6 × 1.5 × 7.4 cm soft tissue mass in the left gluteus maximus muscle (arrows). The mass is hyperintense relative to skeletal muscle T2-weighted fat-suppressed magnetic resonance (MR) image through the left hemipelvis (A) and isointense to skeletal muscle on T1-weighted image (B) with moderately heterogeneous enhancement on post-contrast T1-weighted fat-suppressed image (C). Aggressive MRI features include presence of tumoural necrosis and perilesional oedema/enhancement. There is no extra-compartmental extension
Conclusions
There have been several molecular and genetic advances relating to STT biology incorporated into the most recent WHO classification. The diagnostic accuracy of routine MRI sequences remains less than perfect for the majority of STTs, due to overlapping features, especially after determinate soft tissue masses such as lipoma and ganglia are excluded. A benefit to characterisation has been shown with the use of advanced MRI sequences (DWI, DCE-MRI) [38,39]. There are some STTs that can be diagnosed with high accuracy based on imaging characteristics and location, such as PNSTs (schwannoma and neurofibroma), palmar/plantar fibromatosis, fibromatosis colli, eslatofibroma dorsi, and lipomatosis. In the absence of these determinate entities, the primary role of MRI is to perform local staging of an indeterminate STT rather than provide an accurate histological diagnosis. Solid, indeterminate STTs should be referred to a sarcoma centre for a percutaneous biopsy and ultimately definitive management because a multidisciplinary approach is critical for optimal outcomes for patients with these rare neoplasms. Important imaging features of a particular STT that must be included in a radiological interpretation are the longest lesional dimension, relationship to the superficial fascia (superficial or deep), and the presence of intra-tumoural necrosis, perilesional oedema/enhancement, and extra-compartmental extension. Advanced MRI sequences can be helpful for the presence of hypercellularity (denoted by qualitative restrictive diffusion and quantitatively low ADC values) and neovascularity (visually seen as early arterial enhancement on DCE sequences) suggesting aggressive tumour behaviour. Because the most recent WHO update on STT incorporates tumour biology as an integral feature of STT classification, advanced quantitative MRI sequences will increasingly serve as an important tool in the diagnosis and management of these challenging tumours.