Introduction
Split cord malformation (SCM) is a complex developmental disorder that, in most cases, leads to tethered cord syndrome and the development of neurological and orthopaedic symptoms. The frequency of clinical symptoms increases with the age of the patients, making early diagnosis, proper characterisation of the defect, and the choice of therapeutic strategy, including surgical technique, orthopaedic management, and multi-disciplinary rehabilitation, crucial.
Clinically, SCM, which involves the division of the spinal cord into 2 or more offspring cords, is classified among occult dysraphic defects, which include anomalies of the spinal cord, meninges, and bony structures. The incidence of neurodysraphism in Poland is estimated at 2-3/1000 births, with half being open and closed dysraphism, mainly hernias with a dorsal tumour [1]. Split cord malformation is the most common congenital myelodysplasia, but the lack of a central registry of defects prevents precise determination of its incidence numbers. In a reported larger series of patients, SCM accounted for 20% of all operated neural tube defects and about 5% of all congenital spinal defects [2].
SCM can occur as an isolated defect, which offers a better prognosis, or coexist with other spinal cord and canal defects and central nervous system (CNS) anomalies: hydrocephalus or syringomyelia in 50%, meningeal and meningomyelocele hernias of the undivided spinal cord in 15-25%; herniation of one hemicord in 15-20%, lipomas, dermal sinuses, disontogenetic intrathecal tumours, arachnoid cysts, and tethering adhesions – collectively less than 20%, and Arnold-Chiari malformation, which coexists with SCM in 20%. Patients may also have developmental defects of the gastrointestinal, urogenital, and respiratory systems [3,4].
Manifestations
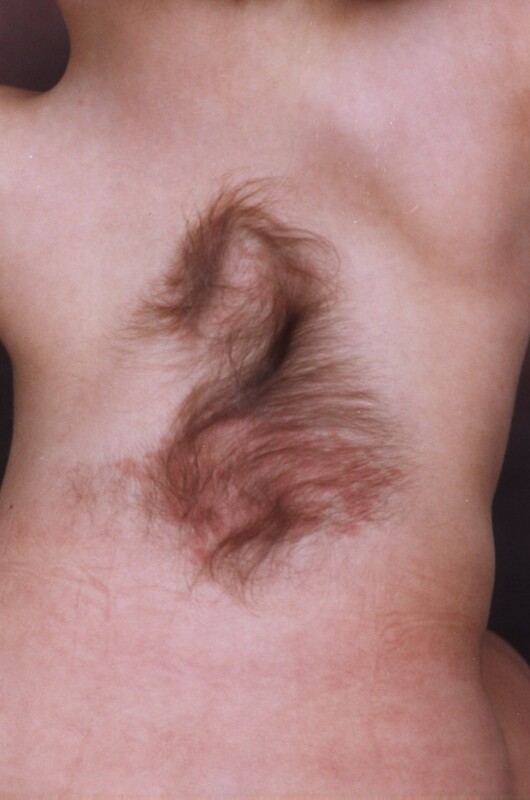
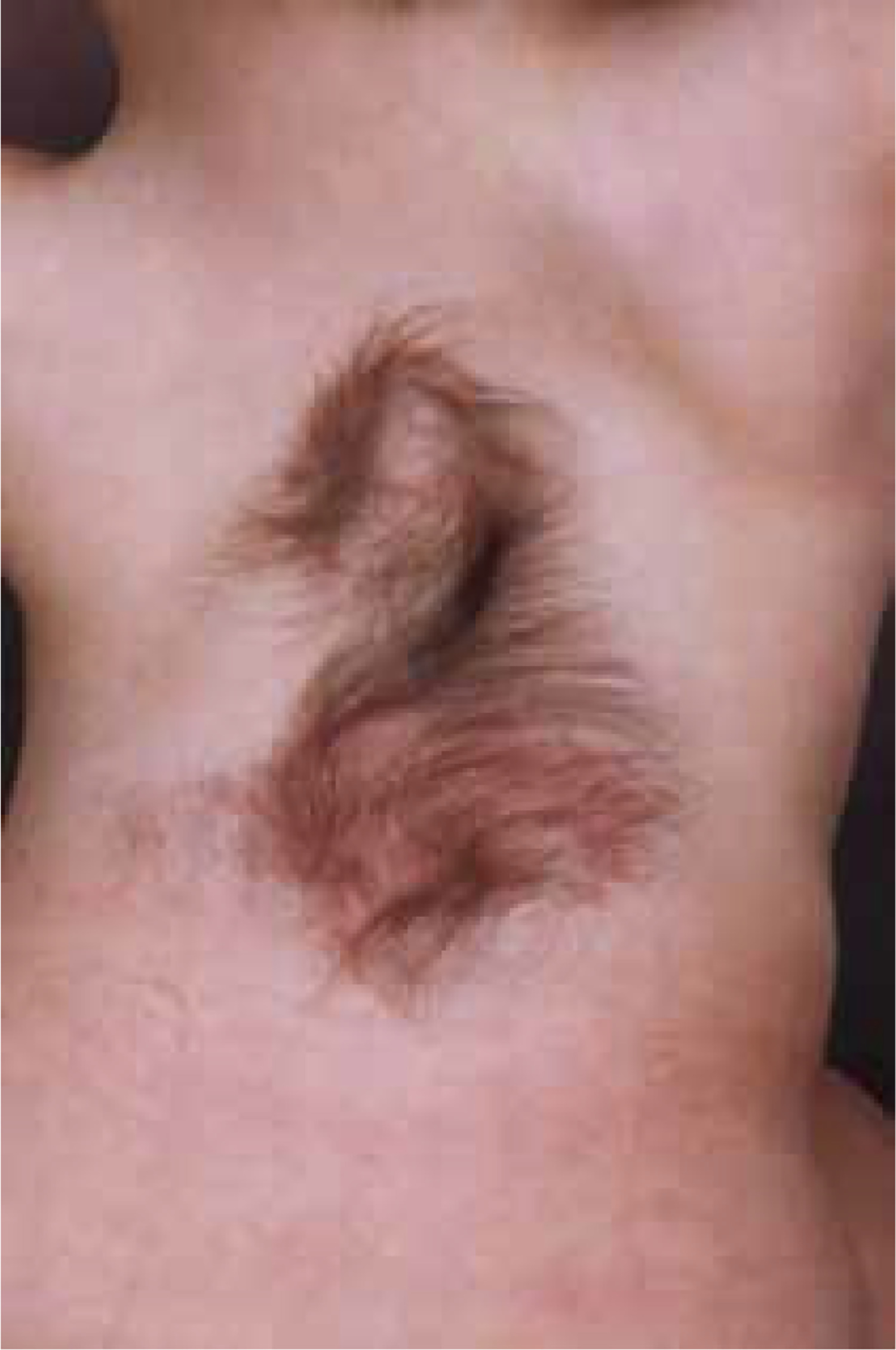
Occult dysraphic defects, when not involving extracanalar expansion, often present with distinct skin changes, spinal bone anomalies, and lower limb abnormalities. Characteristic skin stigmata are found in 50-75% of patients with SCM. These skin alterations, which include tufts of excessive hair, haemangiomas (Figure 1), pigmentation changes, pigmentary nevi, and lipomas, are typically located along the midline of the spine but not necessarily at the exact level of the defect [5]. Structural scoliosis, observed in 80-85% of cases and often necessitating surgical correction [6], is common among these patients. Abnormalities in the lower limbs, such as muscle underdevelopment, limb shortening, clubfoot, and high-arched or flat feet, usually unilaterally, occur in 90% of cases. A specific neuro-orthopaedic syndrome associated with lumbar SCM is the concurrent presence of muscle weakness and atrophy in one lower limb alongside clubfoot, observed in 50% of patients. Hip joint anomalies, including coxa vara and atypical dislocations, are also frequently noted [7,8].
Clinical symptoms of SCM are not distinct and typically emerge during infancy or adolescence, most frequently between the ages of 6 months and 15 years. These symptoms manifest as motor and sensory disturbances in the lower limbs and sphincters, as well as neurotrophic disorders. It is rare for developmental SCM defects to remain undiagnosed in adulthood [9], with the oldest documented case being a 78-year-old woman [10].
Neurological deficits are present in 85-90% of paediatric cases, while adults predominantly experience pain [11,12]. Mental development and intellectual capabilities remain unaffected. Orthopaedic changes and skin manifestations alone do not definitively indicate an SCM defect but should prompt comprehensive neurological examinations and imaging of the entire neural axis to exclude spinal canal and brain abnormalities.
Accurate morphological assessment of the spinal cord, dural sac, fluid spaces, and soft tissues is crucial. This evaluation allows for visualisation of the true defect, identification of cord tethering and its consequences, detection of intrathecal cysts and tumours, and assessment of the vascular plexuses surrounding the spinal canal septum. Identifying and locating concurrent anomalies within the brain and spinal canal can influence the scope and method of the planned surgical intervention and provide prognostic information. Failure to diagnose early and treat surgically in a timely manner may result in irreversible neurological deficits and permanent disability.
Morphology
SCM is characterised by a longitudinal division of the spinal cord into 2 distinct parts within the spinal canal. This segmentation runs parallel to the spine’s long axis and can occur at one or multiple levels, potentially impacting the conus medullaris and filum terminale. The split is demarcated by a septum, which can be oriented sagittally or parasagittally and consists of fibrous, cartilaginous, and/or osseous tissue. Scoliosis may cause rotation of this septum, reorienting the half-canals from a lateral to an anteroposterior configuration. Anteriorly, the bony septum connects to the posterior surface of a vertebral body, while posteriorly it attaches either to the malformed vertebral arches or solely to the dura mater. Fibrous or mixed septa may lack connection to osseous structures and attach only to the dura mater.
The central spinal canal septum anchors the spinal cord in a low position, inhibiting its physiological ascent during spinal growth. In 75-85% of SCM cases, the conus medullaris is positioned below the lower border of the L2 vertebral body [13], with the filum terminale thickened in 50-75% of cases. The spinal cord can be divided asymmetrically into offspring cords, which may involve the entire cord thickness or just the anterior or posterior portions. Partial division is frequently observed in transitional zones adjacent to the fully split region.
In 40-70% of patients, the dura mater and arachnoid are divided into 2 separate sacs, each housing a half-cord. These cases consistently feature a cartilaginous band or osseous spur that traverses the dural and cord splits, linking the vertebral body to posterior canal structures (Figure 2). In 30-60% of patients, the 2 half-cords, each enveloped by its own pia mater, reside within a single dural sac (Figure 3). Each half-cord is supplied by its own anterior spinal artery. This variant lacks a bony spur but includes a fibrous septum penetrating the dura mater [3,6,14], identifiable during surgery or autopsy. Composite SCM, or mixed defects exhibiting characteristics of both primary forms, are seen in 5% of patients [15].
Figure 2
MRI scan, T2-weighted image in the transverse plane. Type I SCM malformation, visible bony septum, and 2 dural sacs, each containing a hemicord
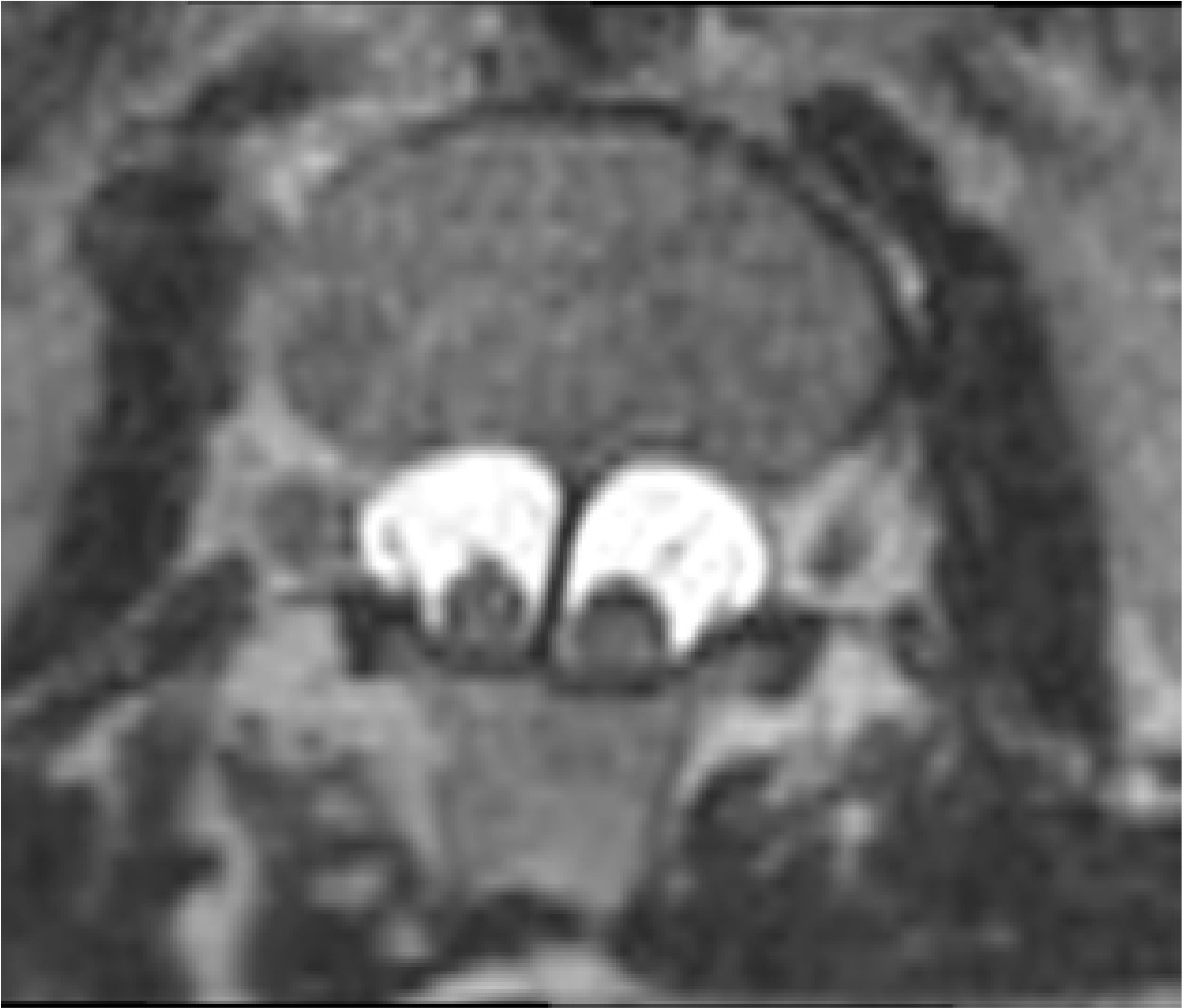
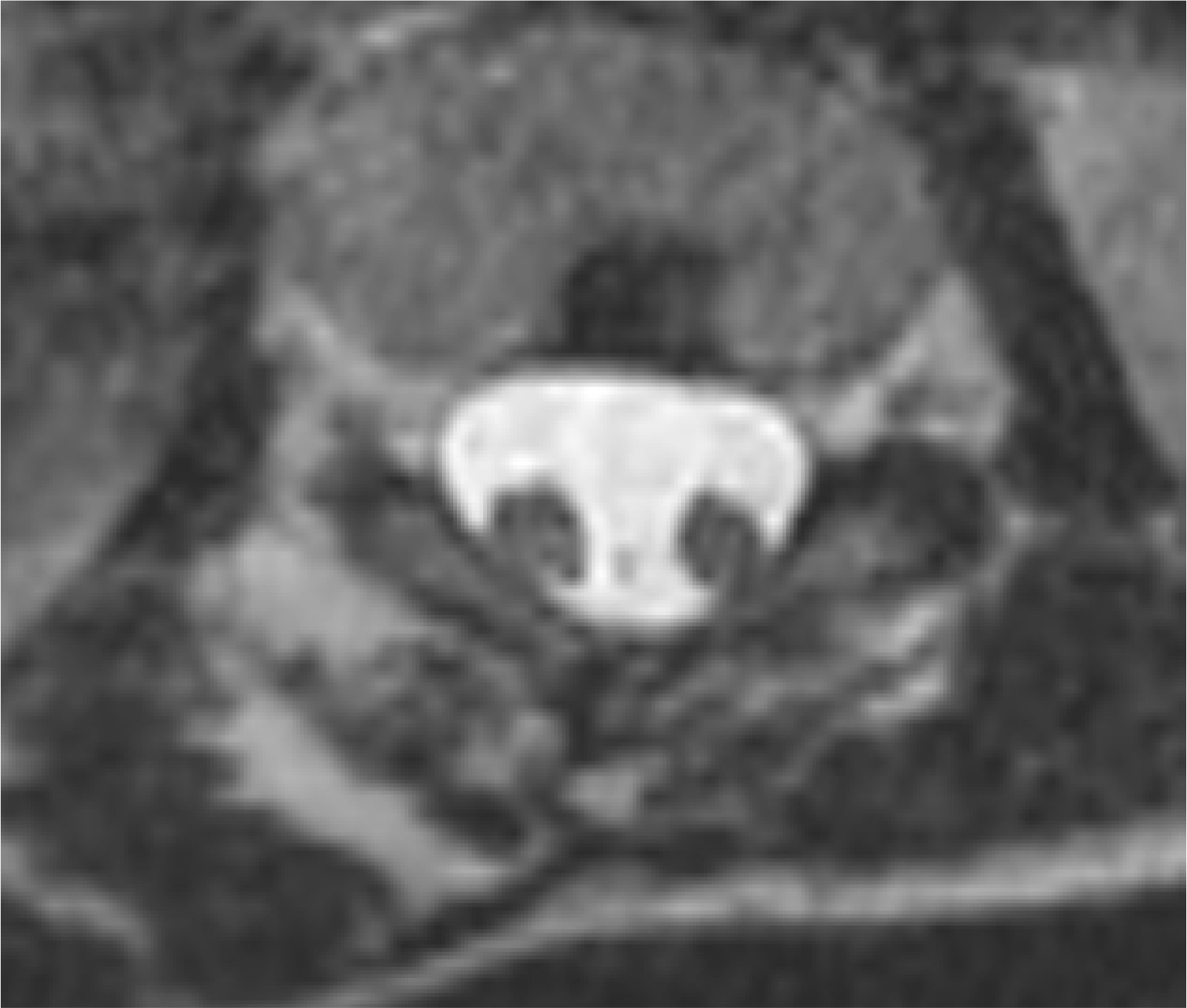
Figure 3
MRI scan, T2-weighted image in the transverse plane. Type II SCM malformation – split spinal cord within a single dural sac
The spine is almost always abnormal. Often exhibited abnormalities include the following: anterior bony clefts in vertebral bodies, posterior clefts in vertebral arches, absent or dysplastic spinous processes, and segmentation or metamerisation defects including hemivertebrae, butterfly vertebrae, wedge-shaped vertebrae, and vertebral body blocks with arch fusions (Figure 4). Laminar fusion and spina bifida, which are pathognomonic for SCM, are present in 60% of cases. Scoliosis or kyphoscoliosis affects over half of SCM patients, while SCM is present in 5-18% of individuals with congenital scoliosis. At the defect level, the spinal canal is abnormally widened, particularly in the coronal plane, indicated by an increased interpedicular distance (Figure 5) [15,16].
Defect location
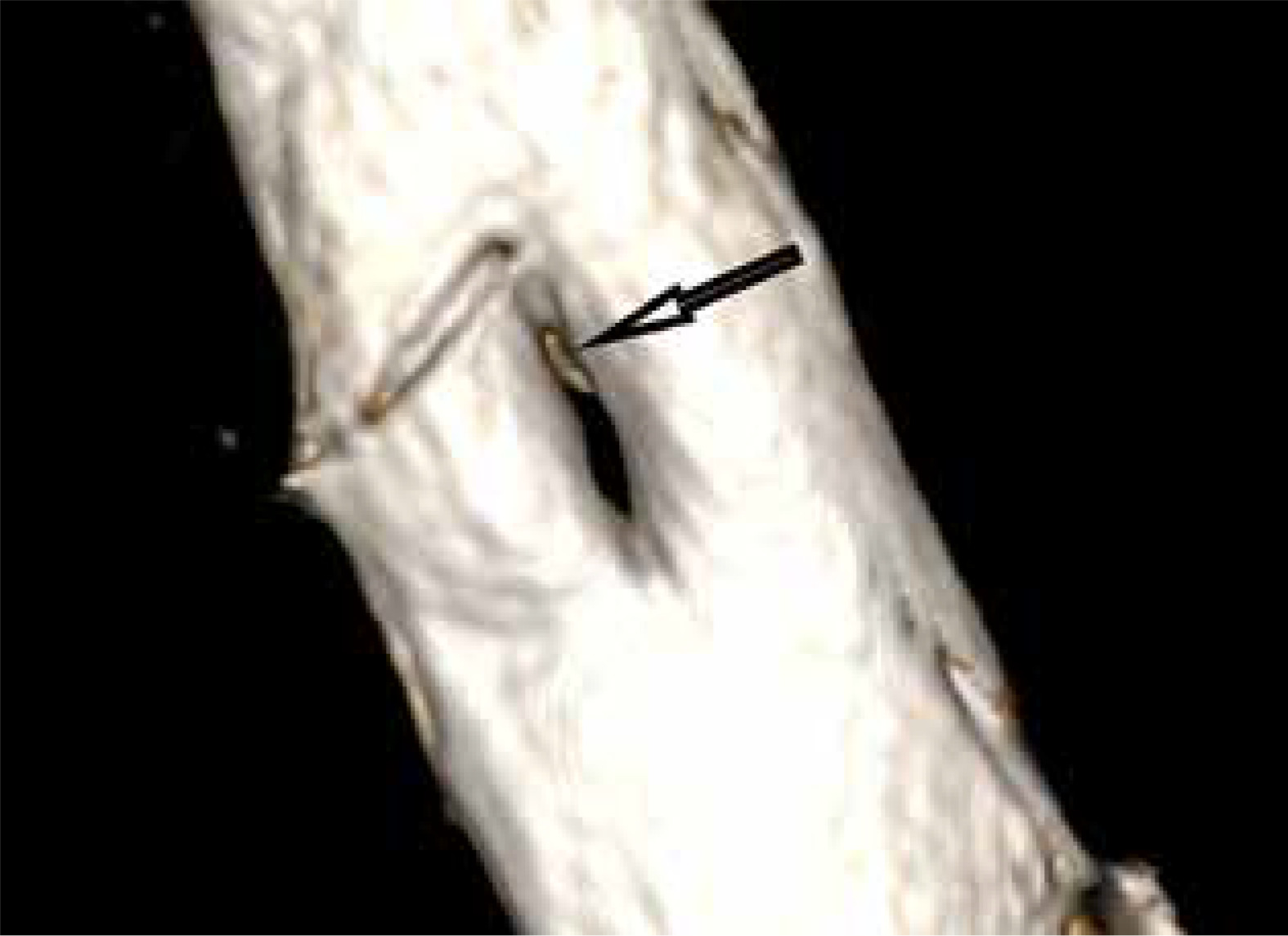
Figure 6
MRI scan, 3D reformatted image from a T2-weighted sequence. A bony spur (arrow) at the upper pole of the split dural sac in a type Ib SCM malformation
The split most commonly occurs at the Th9-S1 levels (81%), in the lumbar region (50%), lower thoracic region (20%), or thoracolumbar region (15-20%). Cervical and upper thoracic splits are less frequently diagnosed (0.8-3%), probably remaining asymptomatic and not causing tethered cord symptoms. Craniovertebral and sub-S1 splits are casuistic [2,17].
Diagnostic imaging
In the diagnostic assessment of the spinal canal and spinal cord, conventional radiographic imaging, as well as advanced imaging techniques such as magnetic resonance imaging (MRI), computed tomography (CT), and ultrasonography (USG), are utilised [18,19].
Conventional radiographs (X-rays) of the spine are a widely available and straightforward method for imaging bony structures. They remain relevant in the initial stages of diagnosing neurodysraphic anomalies, allowing satisfactory evaluation of spinal curvatures and monitoring progression or regression of such changes. X-rays can detect abnormalities related to congenital spinal defects, ribs, and the shoulder and pelvic girdles, thus helping to identify patients requiring further diagnostics and the planning of specialised imaging studies such as MRI and CT.
CT appears to be an ideal method for identifying and evaluating developmental anomalies of the spinal canal. This is due to its very short examination time and the capability for computerised processing of data [20], particularly useful in cases of significant spinal curvatures and deformities. CT has proven to be the best method for assessing bony septa within the spinal canal. Besides providing detailed images of bony structures, it allows the evaluation of soft tissues, both before and after intravenous contrast administration, which is crucial for detecting intrathecal tumours associated with SCM. When performing a CT scan to assess the spine, expanding the field of view or the size of the examined area can also allow for the evaluation of the brain, thorax, or abdomen to identify coexisting anomalies if necessary [19]. However, CT does not provide a comprehensive view of spinal cord disorders.
MRI is the only neuroimaging method that allows for the direct and precise evaluation of the dural sac, spinal cord morphology, conus medullaris, and filum terminale, as well as their associated pathologies. It effectively images the brain, soft tissues inside and outside the spinal canal, and bony structures, making it the preferred modality for assessing a split spinal cord and associated intrathecal damage and tumours [21,22]. MRI can definitively determine the type II SCM. As a non-invasive method, it can be used to screen the entire length of the spinal canal, enabling the detection of complex SCM forms and other spinal cord tethering injuries. MRI is free from artefacts caused by the proximity of bone and air. However, compared to CT, it has a longer examination time and an increased number of motion artefacts. The limitations of MRI are related to the interaction of ferromagnetic materials with the magnetic field. Mechanical and electronic devices, which some patients rely on, may be contraindications for MRI due to potential magnetic field interference. The noise during MRI can cause discomfort, particularly in sensitive patients. Anaesthetic preparation is required for neonates, infants, small children, non-cooperative patients, and those suffering from claustrophobia. MRI is increasingly used for prenatal examinations to evaluate foetuses for developmental defects, including spinal cord and canal dysraphism [23,24].
USG is used to diagnose an intact spinal canal in children because their posterior vertebral elements are not fully ossified. The contents of the spinal canal are best visualised in neonates, who are the most common recipients of spinal USG examinations. Transverse sections better depict cartilaginous and osseous spinal anomalies and adjacent soft tissue pathologies, while longitudinal sections assess the spinal cord, conus medullaris, and cauda equina, as well as intracanalar changes [4]. USG is a dynamic examination, allowing assessment of vascular pulsations and spinal cord and cauda equina mobility within the spinal canal [4,25]. Beyond neonates, pathological spinal changes can also be visualised in infants and young children [26]. The results of USG and MRI correlate well in children assessed for congenital lower spinal defects [27]. Children with features of occult dysraphism should routinely undergo screening examinations [25,27-29]. USG can also be used in prenatal diagnostics [28,29]. For women with a pertinent medical history, targeted foetal USG should be performed at 12-14 weeks of gestation and repeated after 2-3 weeks to confirm the diagnosis [30,31].
Previous terminology
The earliest references to dysraphic spinal cord anomalies appear in the 19th and early 20th centuries. Ollivier from Paris first documented diastematomyelia in 1837. In 1892, Hertwig applied this term to describe the splitting of frog embryos [12]. The term ‘diplomyelia’ was first introduced by Von Recklinghausen in 1886. In 1906, Bruce and colleagues used the term diastematomyelia to describe the splitting of the human spinal cord caused by a bony spur located along the midline of the spinal canal. They reserved the term diplomyelia for cases of true spinal cord duplication without a septum [12].
In 1940, Lichtenstein defined diastematomyelia as a condition in which the spinal cord is divided into halves, each containing a central canal along with one anterior and one posterior horn, and associated roots [32,33]. That same year, Herren and Edwards, based on a series of autopsy studies, defined diplomyelia as either complete or partial duplication of the spinal cord without a spur, where each half of the cord contained a central canal and 2 dorsal and 2 ventral horns [34]. In 1960, Cohen and Sledge questioned the existence of diplomyelia, asserting that the presence of nerve roots emerging from the medial surfaces of each duplicated cord was a necessary diagnostic criterion [12].
The rarity of the condition, combined with inconsistencies among researchers regarding nomenclature, embryological origin, and clinical significance, along with the lack of clearly defined diagnostic criteria, has led to confusion in the neurosurgical literature. Various terms, including diastematomyelia, diplomyelia, dimyelia, hemidydemia, and split cord (hemicord) syndrome, have been used interchangeably or to describe different spinal cord anomalies.
Traditional classification of double spinal cord anomalies based on morphological assessment, particularly through the evaluation of autopsy specimens, seems to linger in clinical practice and the literature [3,33-36]. When each of the 2 spinal cords involved in the anomaly is fully developed, with a central canal and a complete set of 4 horns and corresponding nerve roots, the condition is termed spinal cord duplication – diplomyelia [34,36]. Conversely, if each of the 2 spinal cords is only a half cord, with only one lateral set of ventral and dorsal horns and nerve roots, the condition is described as spinal cord splitting – diastematomyelia [3,33,36].
In 1954, Bremer proposed a new classification system and terminology [35]. This was subsequently revised by Pang and colleagues in 1992, who introduced a unified embryogenetic theory suggesting a common origin for all double cord anomalies stemming from a single ontogenetic error occurring around the time of closure of the primary neuroenteric canal. In 2000, Emura and colleagues confirmed this theory by developing an experimental animal model of the anomaly [37].
Discussion of the new SCM classification
Currently, all defects with divided spinal cords are referred to as SCM, regardless of septum structure. The term diastematomyelia is still used by some authors to refer to type I SCM with a bony septum, while diplomyelia is used for type II SCM. These terms also appear in the literature regarding other SCMs.
The classification proposed by Pang introduced a unified term – SCM – for all cases of duplicated or split spinal cords. The division of SCM into types is based on easily definable characteristics from neuroimaging studies – the presence of a midline septum in the spinal canal and the morphology of the dural sac.
Type I SCM is characterised by the formation of 2 separate dural sacs, each containing its own hemicord, with the medial walls of the sacs enveloping a rigid bony or cartilaginous spur within the spinal canal. Diagnosis of type I SCM is straightforward because the bony septum can be visualised using radiography, CT, or MRI. However, the precise location of the spur relative to the spinal cord split remains contentious [3,12,36]. Pang consistently found the septum at the caudal end of the split during surgery, and Barkovich proposed the identification of a complex type of SCM and the presence of an additional septum when imaging studies reveal a septum at a more rostral level within the cord split. Reports also indicate that the segment of the divided cord can extend several segments below [14].
In 2005, Mahapatra and Gupta proposed a new subclassification of type I SCM, based on the intraoperative location of the bony spur causing the split, which may influence the modification of surgical techniques and postoperative outcomes [16]. They divided the condition into 4 subtypes:
type Ia: a bony spur located in the centre of the split, with the cord divided above and below the spur;
type Ib: a bony spur at the upper pole of the split;
type Ic: a bony spur at the lower pole with a duplicated cord segment above;
type Id: a bony spur extending along the entire length of the split, without cord division above and below the spur.
The most common subtypes observed by the authors were types Ia and Id [16,38]. The risk of hemicord damage is highest in type Id, with authors reporting neurological deterioration post-surgery in 4 out of 6 patients.
In the materials from the Radiology Department of the University Hospital in Kraków, 20 patients (16 children and 4 adults), accounting for 44% of those studied, presented with type I SCM according to the new classification. The septa, composed of spongy bone or delicate compact bone, were identified in 100% of cases using CT and MRI, always extending from the posterior surface of the vertebral body or vertebral block to the corresponding arches, dividing the spinal canal into more or less symmetrical parts. The duplication of the dural sac was consistently visible on MRI and corresponded to the level of the septum. The type I septum was consistently located at the caudal end of the spinal cord split [6], categorising these cases as subtype Ic in the Mahapatra classification. These observations are consistent with the findings from Pang’s series [15], and they have been confirmed in the authors’ subsequent clinical experiences.
Type II SCM features hemicords running within a single, shared dural sac. Each hemicord has its own anterior spinal artery. There is no rigid bony spur, and the midline septum is composed of fibrous or fibrous-vascular tissue. In the series of diagnostic imaging examinations analysed in Kraków, this type of anomaly was found in 36% of cases – 16 patients (14 children and 2 adults). Both hemicords, more frequently symmetrical, lie close together within a single, broad dural sac. In contrast to type I, where the bony septum was easily visible on imaging studies, the fibrous septum in type II was detected in only one patient via MRI [6]. This fact is not surprising in light of the literature, as most authors emphasise the limited efficacy of available diagnostic methods in identifying this aspect of the anomaly [15,24,36]. Pang visualised fibrous septa in MRI in only 3 out of 18 patients, while Ersahin did not detect it in any of the 18 patients with type II anomalies in his group. However, both authors consistently reported the presence of the septum in all patients during surgery. All fibrous septa were found at the distal end of the spinal cord split and caused its tethering to the dural sac. The spinal attachment was characteristically always found at a higher level than the dural attachment.
MRI accurately visualised the spinal cord split and the fluid space of the dural sac in 100% of cases [3,36] therefore it can be considered the method of choice for the diagnosis of type II SCM.
Complex forms of the anomaly encompass cases with multiple spinal canal septa, exhibiting diverse structures and morphologies. The term type III SCM, also known as composite SCM, is designated for multiple, non-contiguous SCMs located at different spinal levels [39]. These lesions may present the same type of anomaly or a combination of types, with septa of bony and/or fibrous composition (Figure 7). The literature documents very few cases of composite SCM.
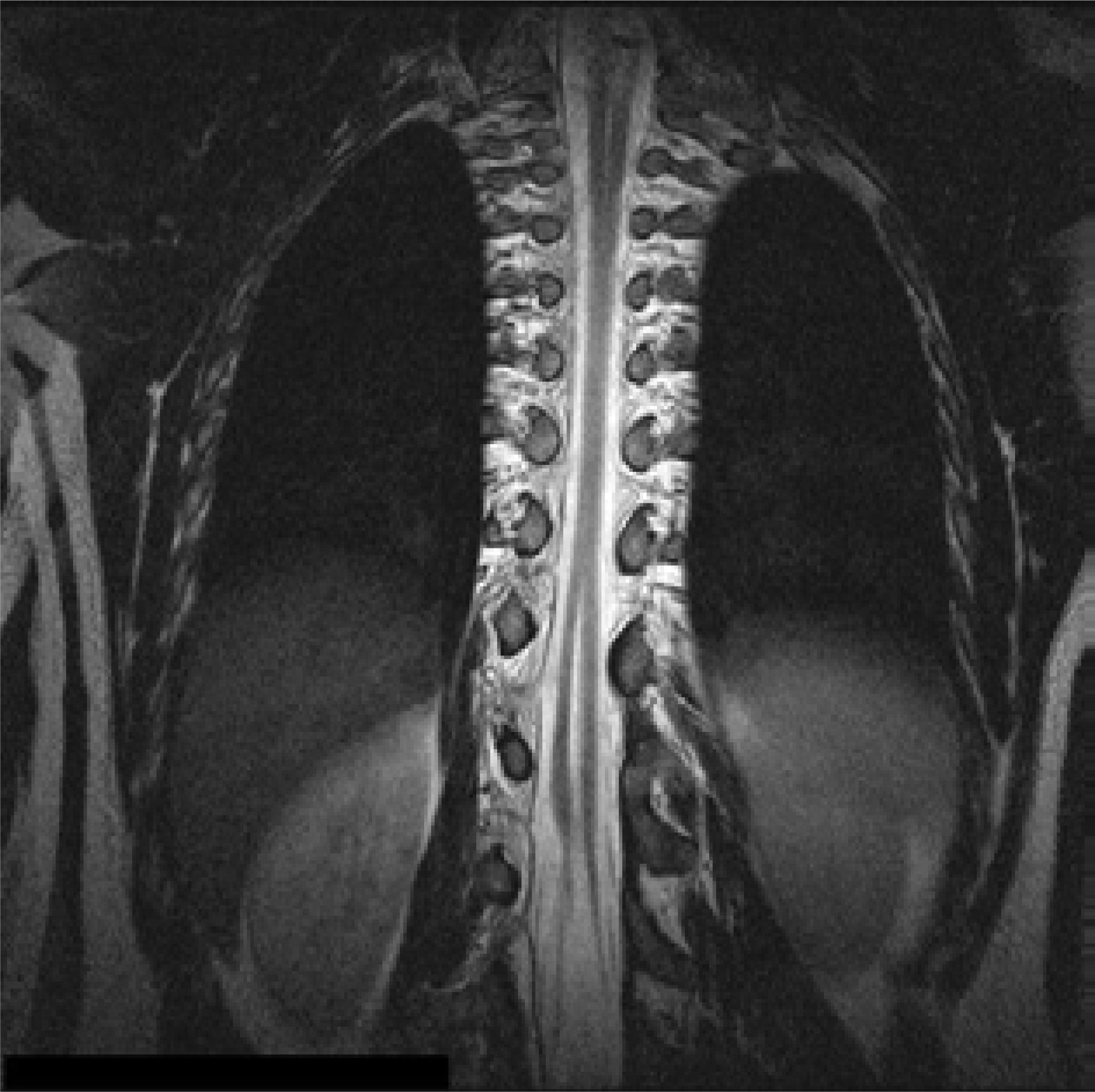
Figure 7
MRI scan, T2-weighted image in the coronal plane. SCM type III. Type I with a bony septum in the cervical region and type II in the thoracolumbar region, separated by a long segment of a normal spinal cord
In 2019, Meena and Doddamani described an atypical, unique variant of mixed SCM that included features of both classical types of SCM at the same level, with a central septum of osseous-fibrous composition, and proposed the designation of type 1.5 SCM. Dorsally located, incomplete bony spurs represented subtype 1.5 A, while bony spurs in the ventral part of the spinal canal represented subtype 1.5 B (Figure 8). To date, the literature has recorded 15 cases, with one-third being the Meena and Doddamani’s patients [40]. Type 1.5 B remains a surgical challenge compared to the relatively easier-to-excise subtype 1.5 A. The term 1.5 SCM remains a topic of debate among researchers [41]. Some express reservations about incorporating this nomenclature into the general classification, highlighting the intraoperatively confirmed possibility of an incomplete bony spur without a fibrous component [41]. Consequently, they propose the term SCM 0.5. Moreover, these researchers question the possibility of formation of a septum composed of both bony and fibrous components, considering them embryologically distinct structures. They explain the emergence of the 0.5 variant as a result of the regression of a complete bony septum [41]. However, there are also voices supporting the introduction of type 1.5 along with pathogenetic theories [42].
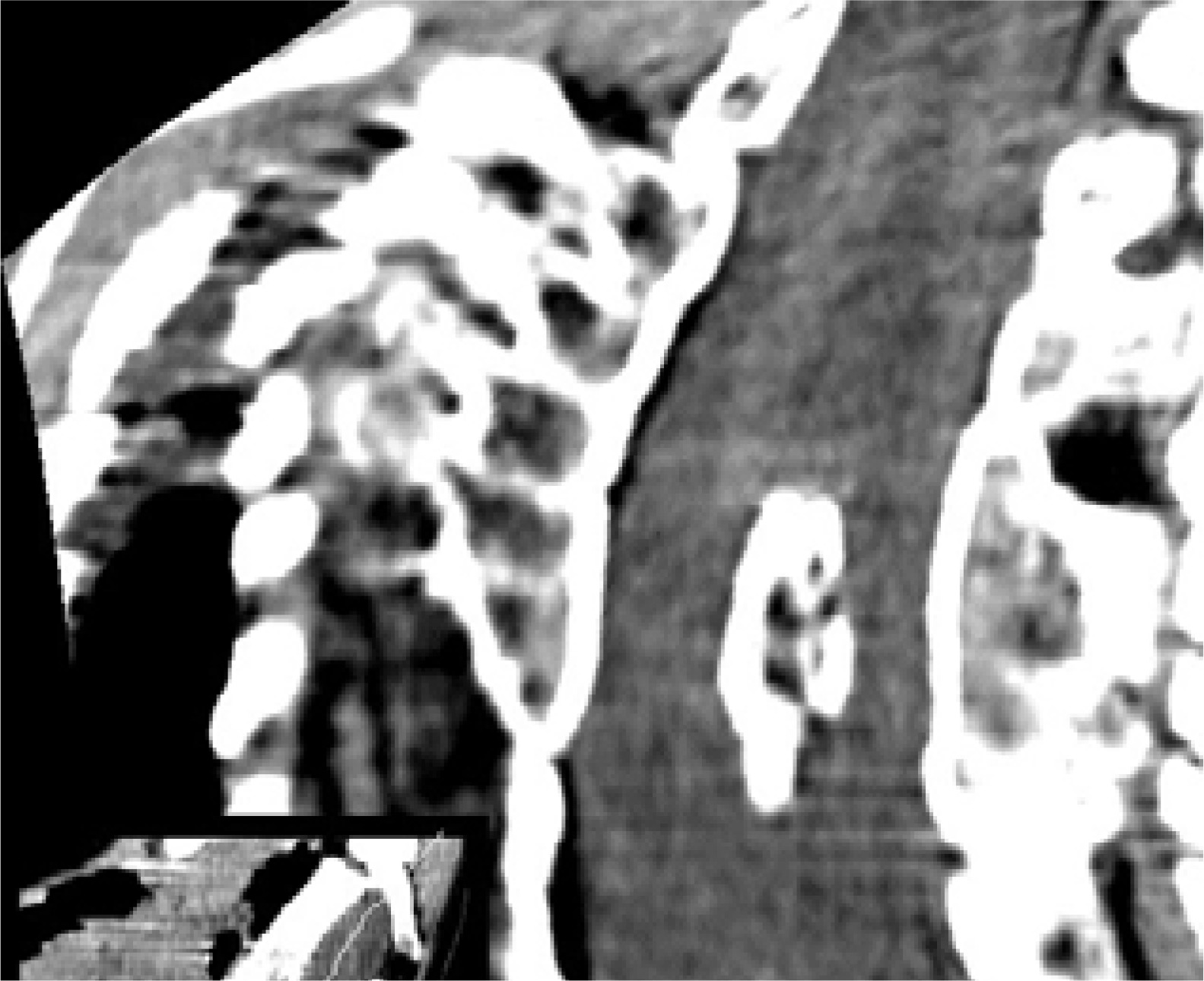
Figure 8
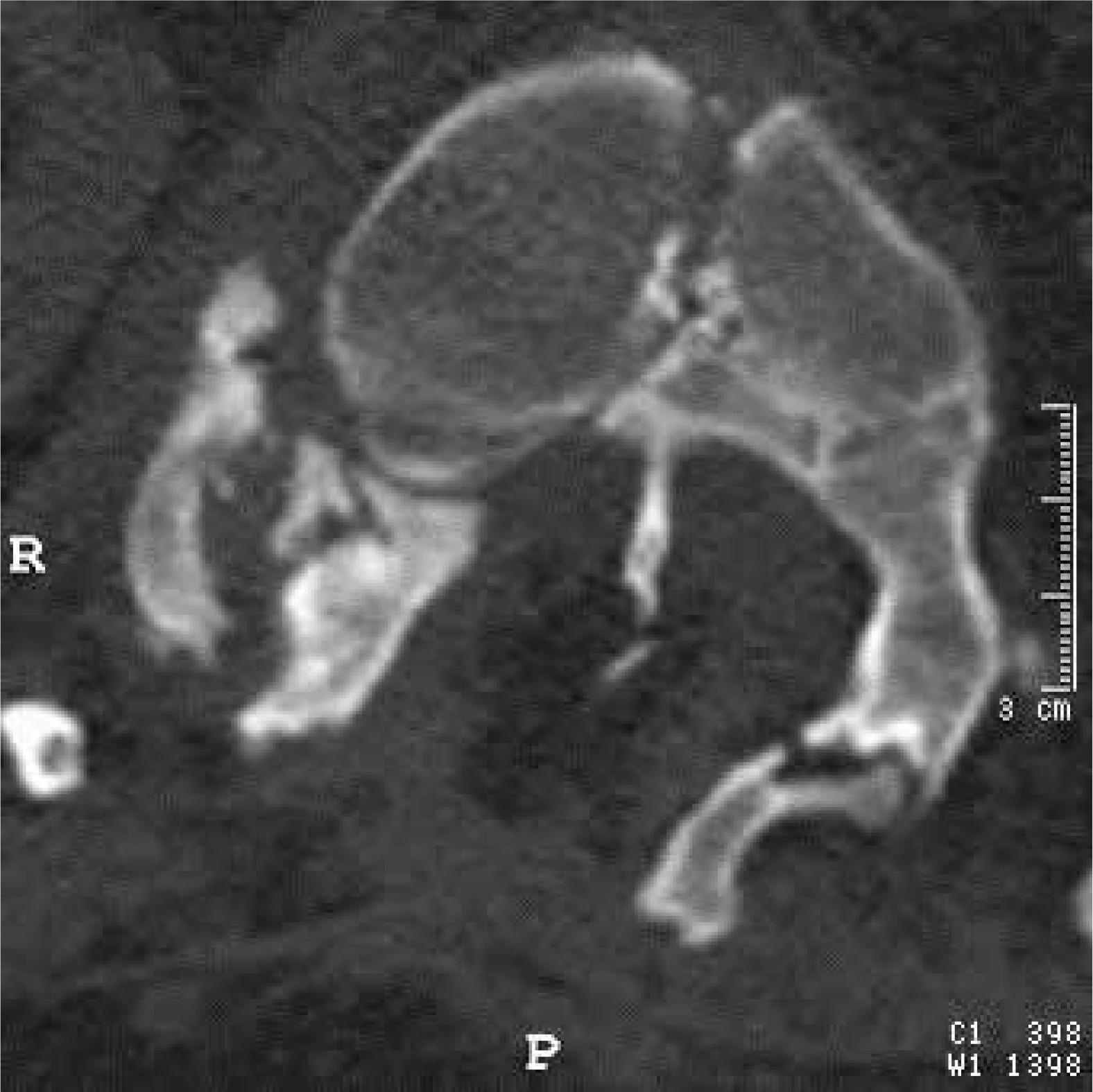
CT scan, transverse section at the level of the bony septum of the spinal canal; mixed SCM variant, subtype 1.5B – a bony spur in the ventral part of the spinal canal, with the dorsal end of the spur embedded in fatty tissue
In Krakow’s series [6], multiple bony septa were identified in 9 patients. Multiple type I SCM was found in 3 patients; two had 2 and one had 3 bony septa arranged one after the other within one common spinal cord split. Two patients exhibited a composite lesion consisting of a single type I and a single type II (Figures 9 and 10), while one patient had a type I bony septum alongside Doddamani’s type 1.5 SCM – a mixed osseous-fibrous (Figure 11). In these patients, both septa were located within a single segment of the spinal cord split, with type I being more cranial relative to type II. In one case, 2 bony spurs were located on adjacent spinal segments, separated by type II, with a fibrous septum visible on MRI.
Figure 9
MRI scan, 3-plane MPR from a T2-weighted sequence, conducted through the bony septum of the spinal canal. Complex SCM. Type I at the L3 level – short segmental duplication of the dural sac visible in transverse images and the bony septum visible in all 3 planes. The split cord is visible above the septum up to the Th12 level
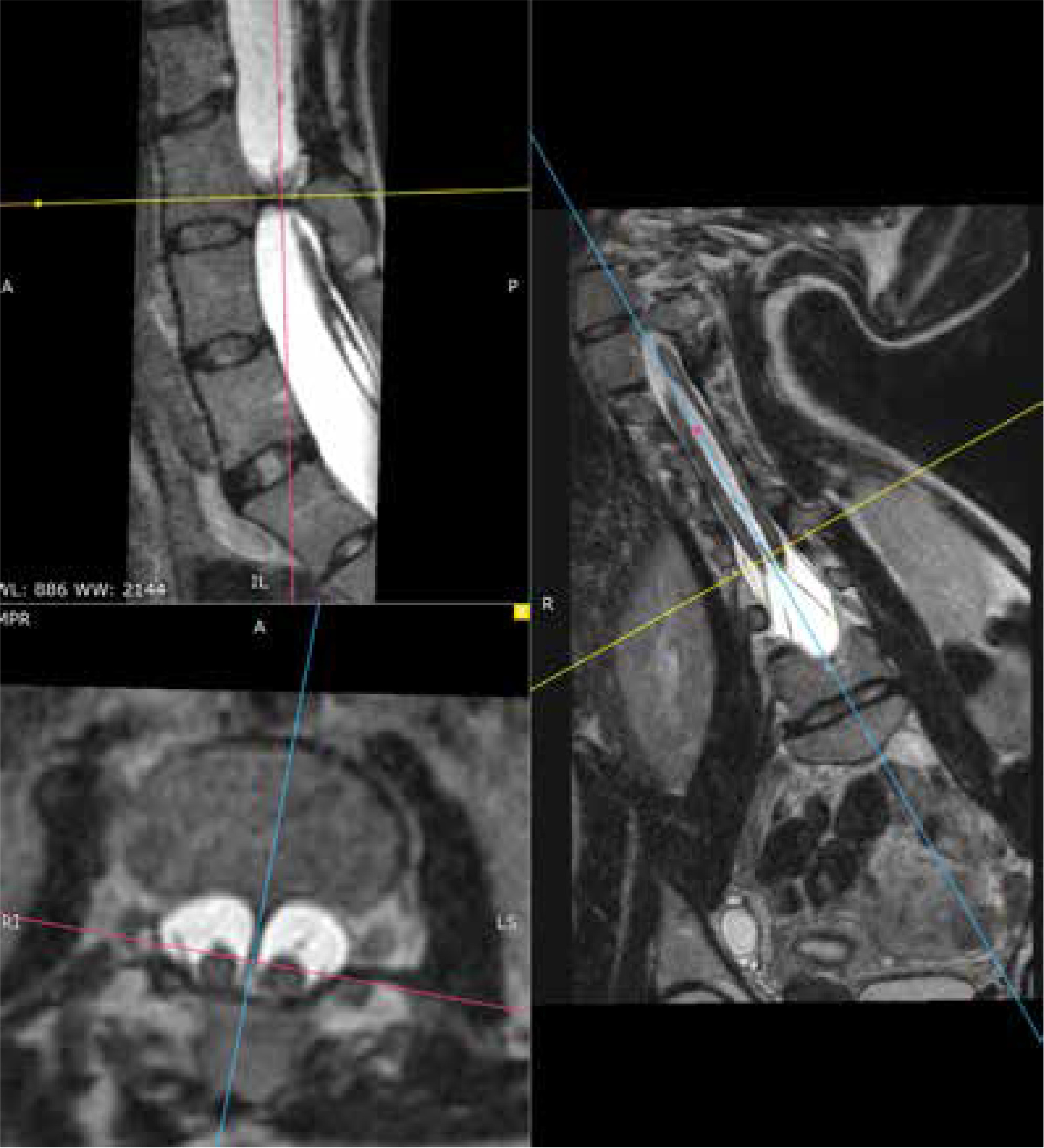
Figure 10
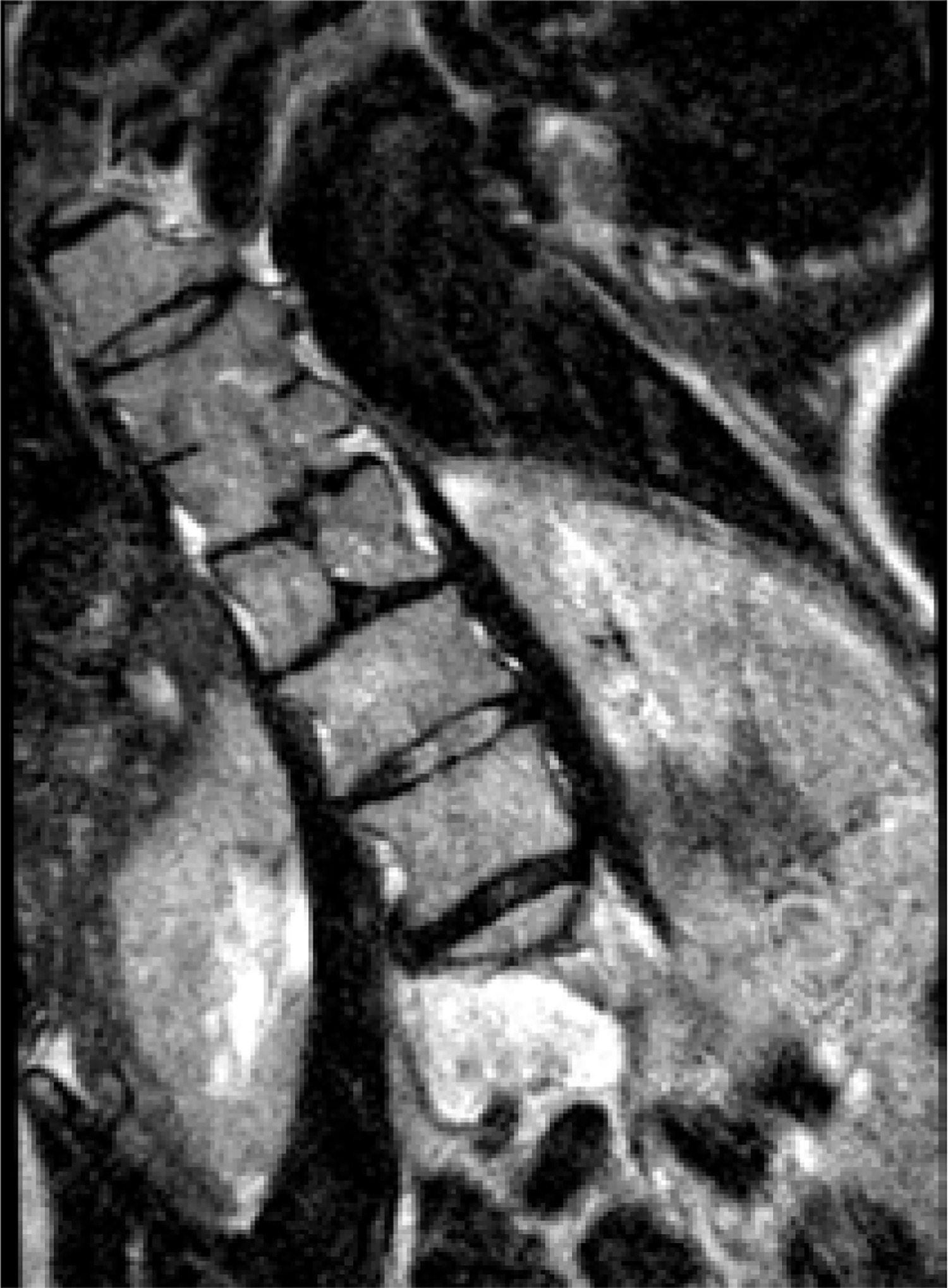
MRI scan, 3-plane MPR from a T2-weighted sequence, conducted below the bony septum of the spinal canal. Complex SCM. Type II malformation – 2 hemicords within a single dural sac, with a single conus located at the S1 level; a hydrosyringomyelic cavity is present within the left hemicord
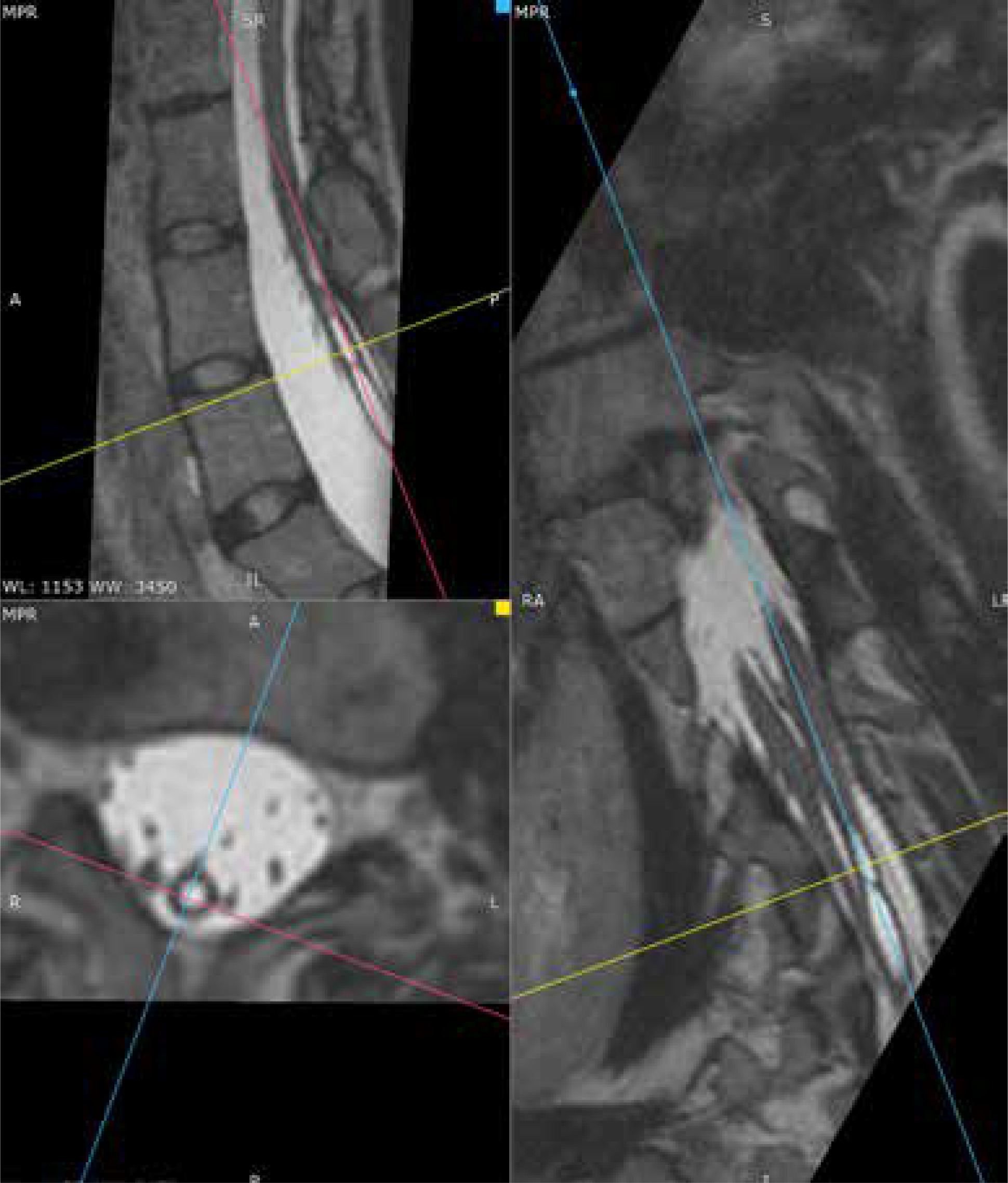
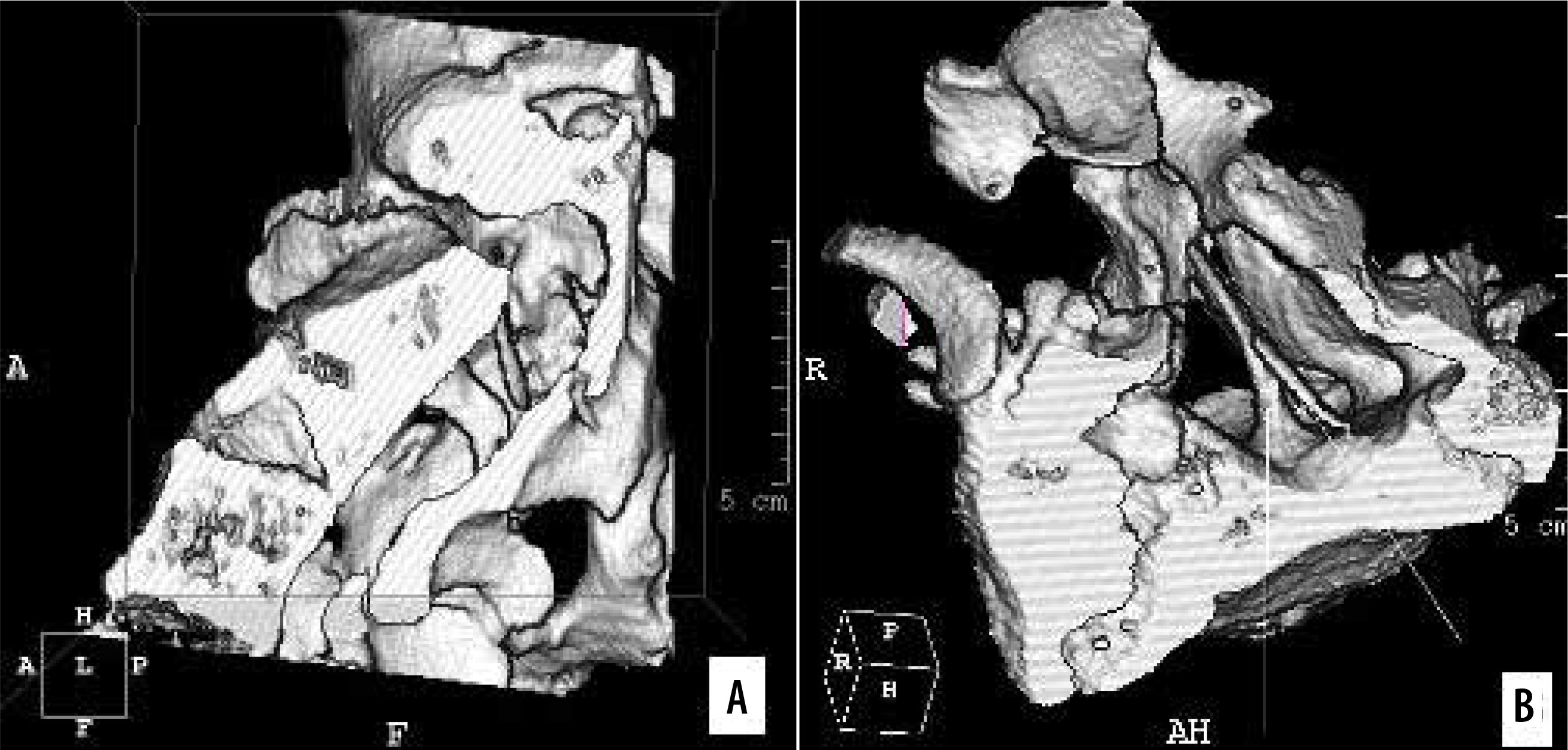
Figure 11
CT scan, 3D reformatted image (SSD). Complex SCM type I/1.5B. Two bony septa at adjacent spinal levels. A) Axial section of the spinal canal. B) Transverse section, top-down view. The sagittal bony septum traverses the entire canal (type I), and the incomplete septum connects to the ventral boundary of the spinal canal (type 1.5B), marked with arrows
The last 2 patients in this group exhibited different morphologies of a lesion comprising a single type I and a single type II, but the septa had separate, unconnected spinal splits, fulfilling the criteria for type III SCM. In this instance, type II was found at a lower spinal level. The segment of normal spinal cord separating the multiple lesions could vary in length, ranging from one to several spinal segments in the analysed material.
The least frequently reported example of a composite lesion is the tripartite spinal cord. This type of lesion was described by Podsiadło-Kleinrok [6] in a 12-year-old girl. Two spinal canal septa were located at levels Th9 and Th12-L1. The higher bony spur divided the cord into 2 symmetrical parts; below this, the right hemicord exhibited an additional division at an obliquely oriented incomplete bony septum, attaching to the vertebral arch and not traversing the entire width of the spinal canal lumen. Pang also reports a similar case in his series [12]. Complex forms of SCM appear to be the most challenging to classify, with no standardised nomenclature yet established and a lack of consensus regarding their classification.
Conclusions
In the imaging of the spinal cord, conus medullaris, filum terminale, and dural sac, MRI has proven to be the most effective, making it the method of choice for detecting SCM defects. CT is the method of choice for assessing bony structures and is most useful for defining single and multiple bony and osteochondral septa. It also visualises spinal bony anomalies, making it indispensable for planning the correction of spinal deformities. It is advisable that all patients suspected of having SCM based on one imaging modality also undergo evaluation using the other imaging technique.
Confirmation and categorisation of the defect rely on the assessment of the dural sac and the bony septum of the canal accompanying the spinal cord splitting. A bony canal septum and a double dural sac are sufficient to diagnose type I SCM, while the absence of a bony septum and a single dural sac determine the classification of the defect as type II. Other morphological forms of the defect are also possible, which can be described as the coexistence of at least 2 changes, each describable as type I or II. A uniform classification of such defects is still lacking. An exceptional case is the composite type, in which more than one segment of the split cord is observed.
Regardless of the defect type, every SCM is a tethering lesion of the spinal cord, and the risk of neurological deficits in SCM patients increases with age. Indications for diagnostic imaging should include the presence of an open neural tube defect, spinal deformity, abnormalities in the lower limbs, or skin stigmata of occult neurodysraphism. Such examinations should especially be performed before planned spinal deformity correction surgeries.
A comprehensive examination of the entire neural axis is essential for the detection of multiple instances of split cord malformation, accompanying tethering lesions, intramedullary foci, and intracanal tumours.
Preoperative classification of the defect is crucial because surgical techniques differ for each type of SCM. Early surgical intervention in all symptomatic patients with SCM may be associated with better postoperative outcomes. Prophylactic surgical interventions are recommended as well for asymptomatic patients.